Look up de novo in Wiktionary, the free dictionary.
In general usage, de novo (literally 'of new') is a Latin expression used in English to mean 'from the beginning', 'anew'.
De novo may also refer to:
In general usage, de novo (literally 'of new') is a Latin expression used in English to mean 'from the beginning', 'anew'.
De novo may also refer to:

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

Protein biosynthesis is a core biological process, occurring inside cells, balancing the loss of cellular proteins through the production of new proteins. Proteins perform a number of critical functions as enzymes, structural proteins or hormones. Protein synthesis is a very similar process for both prokaryotes and eukaryotes but there are some distinct differences.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.
The coding region of a gene, also known as the coding sequence (CDS), is the portion of a gene's DNA or RNA that codes for a protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

Top7 is an artificial protein, classified as a de novo protein. This means that the protein itself was designed to have a specific structure and functional properties.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "candidate-gene" approach.
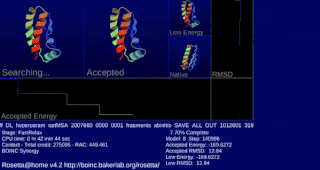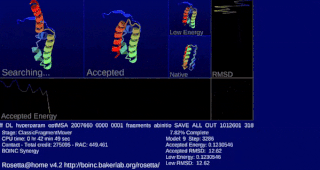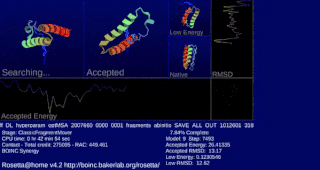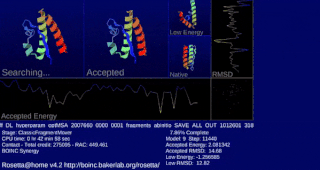
Rosetta@home is a volunteer computing project researching protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker lab. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.
In biology, the word gene has two meanings. The Mendelian gene is a basic unit of heredity. The molecular gene is a sequence of nucleotides in DNA, that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and non-coding genes.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.
The 1000 Plant Transcriptomes Initiative (1KP) was an international research effort to establish the most detailed catalogue of genetic variation in plants. It was announced in 2008 and headed by Gane Ka-Shu Wong and Michael Deyholos of the University of Alberta. The project successfully sequenced the transcriptomes of 1000 different plant species by 2014; its final capstone products were published in 2019.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
In molecular phylogenetics, relationships among individuals are determined using character traits, such as DNA, RNA or protein, which may be obtained using a variety of sequencing technologies. High-throughput next-generation sequencing has become a popular technique in transcriptomics, which represent a snapshot of gene expression. In eukaryotes, making phylogenetic inferences using RNA is complicated by alternative splicing, which produces multiple transcripts from a single gene. As such, a variety of approaches may be used to improve phylogenetic inference using transcriptomic data obtained from RNA-Seq and processed using computational phylogenetics.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.
The Comprehensive Antibiotic Resistance Database (CARD) is a biological database that collects and organizes reference information on antimicrobial resistance genes, proteins and phenotypes. The database covers all types of drug classes and resistance mechanisms and structures its data based on an ontology. The CARD database was one of the first resources that covered antimicrobial resistance genes. The resource is updated monthly and provides tools to allow users to find potential antibiotic resistance genes in newly-sequenced genomes.