In chemical analysis, chromatography is a laboratory technique for the separation of a mixture into its components. The mixture is dissolved in a fluid solvent called the mobile phase, which carries it through a system on which a material called the stationary phase is fixed. Because the different constituents of the mixture tend to have different affinities for the stationary phase and are retained for different lengths of time depending on their interactions with its surface sites, the constituents travel at different apparent velocities in the mobile fluid, causing them to separate. The separation is based on the differential partitioning between the mobile and the stationary phases. Subtle differences in a compound's partition coefficient result in differential retention on the stationary phase and thus affect the separation.

In biochemistry, denaturation is a process in which proteins or nucleic acids lose the quaternary structure, tertiary structure, and secondary structure which is present in their native state, by application of some external stress or compound, such as a strong acid or base, a concentrated inorganic salt, an organic solvent, agitation and radiation, or heat. If proteins in a living cell are denatured, this results in disruption of cell activity and possibly cell death. Protein denaturation is also a consequence of cell death. Denatured proteins can exhibit a wide range of characteristics, from conformational change and loss of solubility or dissociation of cofactors to aggregation due to the exposure of hydrophobic groups. The loss of solubility as a result of denaturation is called coagulation. Denatured proteins lose their 3D structure, and therefore, cannot function.

The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.
In molecular biology, restriction fragment length polymorphism (RFLP) is a technique that exploits variations in homologous DNA sequences, known as polymorphisms, populations, or species or to pinpoint the locations of genes within a sequence. The term may refer to a polymorphism itself, as detected through the differing locations of restriction enzyme sites, or to a related laboratory technique by which such differences can be illustrated. In RFLP analysis, a DNA sample is digested into fragments by one or more restriction enzymes, and the resulting restriction fragments are then separated by gel electrophoresis according to their size.

High-performance liquid chromatography (HPLC), formerly referred to as high-pressure liquid chromatography, is a technique in analytical chemistry used to separate, identify, and quantify specific components in mixtures. The mixtures can originate from food, chemicals, pharmaceuticals, biological, environmental and agriculture, etc., which have been dissolved into liquid solutions.
The first isolation of deoxyribonucleic acid (DNA) was done in 1869 by Friedrich Miescher. DNA extraction is the process of isolating DNA from the cells of an organism isolated from a sample, typically a biological sample such as blood, saliva, or tissue. It involves breaking open the cells, removing proteins and other contaminants, and purifying the DNA so that it is free of other cellular components. The purified DNA can then be used for downstream applications such as PCR, sequencing, or cloning. Currently, it is a routine procedure in molecular biology or forensic analyses.

Temperature gradient gel electrophoresis (TGGE) and denaturing gradient gel electrophoresis (DGGE) are forms of electrophoresis which use either a temperature or chemical gradient to denature the sample as it moves across an acrylamide gel. TGGE and DGGE can be applied to nucleic acids such as DNA and RNA, and proteins. TGGE relies on temperature dependent changes in structure to separate nucleic acids. DGGE separates genes of the same size based on their different denaturing ability which is determined by their base pair sequence. DGGE was the original technique, and TGGE a refinement of it.
The overlap extension polymerase chain reaction is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used assemble multiple smaller double stranded DNA fragments into a larger DNA sequence. OE-PCR is widely used to insert mutations at specific points in a sequence or to assemble custom DNA sequence from smaller DNA fragments into a larger polynucleotide.

Heteroduplex analysis (HDA) is a method in biochemistry used to detect point mutations in DNA since 1992. Heteroduplexes are dsDNA molecules that have one or more mismatched pairs, on the other hand homoduplexes are dsDNA which are perfectly paired. This method of analysis depend up on the fact that heteroduplexes shows reduced mobility relative to the homoduplex DNA. heteroduplexes are formed between different DNA alleles. In a mixture of wild-type and mutant amplified DNA, heteroduplexes are formed in mutant alleles and homoduplexes are formed in wild-type alleles. There are two types of heteroduplexes based on type and extent of mutation in the DNA. Small deletions or insertion create bulge-type heteroduplexes which is stable and is verified by electron microscope. Single base substitutions creates more unstable heteroduplexes called bubble-type heteroduplexes, because of low stability it is difficult to visualize in electron microscopy. HDA is widely used for rapid screening of mutation of the 3 bp p.F508del deletion in the CFTR gene.
Reversed-phase liquid chromatography (RP-LC) is a mode of liquid chromatography in which non-polar stationary phase and polar mobile phases are used for the separation of organic compounds. The vast majority of separations and analyses using high-performance liquid chromatography (HPLC) in recent years are done using the reversed phase mode. In the reversed phase mode, the sample components are retained in the system the more hydrophobic they are.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
Multiplex ligation-dependent probe amplification (MLPA) is a variation of the multiplex polymerase chain reaction that permits amplification of multiple targets with only a single primer pair. It detects copy number changes at the molecular level, and software programs are used for analysis. Identification of deletions or duplications can indicate pathogenic mutations, thus MLPA is an important diagnostic tool used in clinical pathology laboratories worldwide.

Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
An allele-specific oligonucleotide (ASO) is a short piece of synthetic DNA complementary to the sequence of a variable target DNA. It acts as a probe for the presence of the target in a Southern blot assay or, more commonly, in the simpler dot blot assay. It is a common tool used in genetic testing, forensics, and molecular biology research.
Melting curve analysis is an assessment of the dissociation characteristics of double-stranded DNA during heating. As the temperature is raised, the double strand begins to dissociate leading to a rise in the absorbance intensity, hyperchromicity. The temperature at which 50% of DNA is denatured is known as the melting temperature. Measurement of melting temperature can help us predict species by just studying the melting temperature. This is because every organism has a specific melting curve.
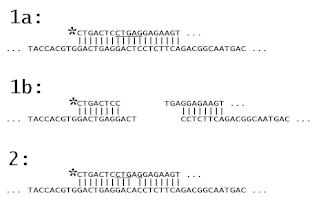
Oligomer Restriction is a procedure to detect an altered DNA sequence in a genome. A labeled oligonucleotide probe is hybridized to a target DNA, and then treated with a restriction enzyme. If the probe exactly matches the target, the restriction enzyme will cleave the probe, changing its size. If, however, the target DNA does not exactly match the probe, the restriction enzyme will have no effect on the length of the probe. The OR technique, now rarely performed, was closely associated with the development of the popular polymerase chain reaction (PCR) method.
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
COLD-PCR is a modified polymerase chain reaction (PCR) protocol that enriches variant alleles from a mixture of wildtype and mutation-containing DNA. The ability to preferentially amplify and identify minority alleles and low-level somatic DNA mutations in the presence of excess wildtype alleles is useful for the detection of mutations. Detection of mutations is important in the case of early cancer detection from tissue biopsies and body fluids such as blood plasma or serum, assessment of residual disease after surgery or chemotherapy, disease staging and molecular profiling for prognosis or tailoring therapy to individual patients, and monitoring of therapy outcome and cancer remission or relapse. Common PCR will amplify both the major (wildtype) and minor (mutant) alleles with the same efficiency, occluding the ability to easily detect the presence of low-level mutations. The capacity to detect a mutation in a mixture of variant/wildtype DNA is valuable because this mixture of variant DNAs can occur when provided with a heterogeneous sample – as is often the case with cancer biopsies. Currently, traditional PCR is used in tandem with a number of different downstream assays for genotyping or the detection of somatic mutations. These can include the use of amplified DNA for RFLP analysis, MALDI-TOF genotyping, or direct sequencing for detection of mutations by Sanger sequencing or pyrosequencing. Replacing traditional PCR with COLD-PCR for these downstream assays will increase the reliability in detecting mutations from mixed samples, including tumors and body fluids.
Multiple Annealing and Looping Based Amplification Cycles (MALBAC) is a quasilinear whole genome amplification method. Unlike conventional DNA amplification methods that are non-linear or exponential, MALBAC utilizes special primers that allow amplicons to have complementary ends and therefore to loop, preventing DNA from being copied exponentially. This results in amplification of only the original genomic DNA and therefore reduces amplification bias. MALBAC is “used to create overlapped shotgun amplicons covering most of the genome”. For next generation sequencing, MALBAC is followed by regular PCR which is used to further amplify amplicons.

Surveyor nuclease assay is an enzyme mismatch cleavage assay used to detect single base mismatches or small insertions or deletions (indels).