Genetic drift, also known as random genetic drift, allelic drift or the Wright effect, is the change in the frequency of an existing gene variant (allele) in a population due to random chance.
Small populations can behave differently from larger populations. They are often the result of population bottlenecks from larger populations, leading to loss of heterozygosity and reduced genetic diversity and loss or fixation of alleles and shifts in allele frequencies. A small population is then more susceptible to demographic and genetic stochastic events, which can impact the long-term survival of the population. Therefore, small populations are often considered at risk of endangerment or extinction, and are often of conservation concern.

The neutral theory of molecular evolution holds that most evolutionary changes occur at the molecular level, and most of the variation within and between species are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin.
Fitness is a quantitative representation of individual reproductive success. It is also equal to the average contribution to the gene pool of the next generation, made by the same individuals of the specified genotype or phenotype. Fitness can be defined either with respect to a genotype or to a phenotype in a given environment or time. The fitness of a genotype is manifested through its phenotype, which is also affected by the developmental environment. The fitness of a given phenotype can also be different in different selective environments.
Population genetics is a subfield of genetics that deals with genetic differences within and among populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.
Allele frequency, or gene frequency, is the relative frequency of an allele at a particular locus in a population, expressed as a fraction or percentage. Specifically, it is the fraction of all chromosomes in the population that carry that allele over the total population or sample size. Microevolution is the change in allele frequencies that occurs over time within a population.
The effective population size (Ne) is the size of an idealised population that would experience the same rate of genetic drift as the real population. The effective population size is normally smaller than the census population size N, partly because chance events prevent some individuals from breeding, and partly due to background selection and genetic hitchhiking. Idealised populations are based on unrealistic but convenient assumptions including random mating, rarity of natural selection such that each gene evolves independently, and constant population size.
Genetic load is the difference between the fitness of an average genotype in a population and the fitness of some reference genotype, which may be either the best present in a population, or may be the theoretically optimal genotype. The average individual taken from a population with a low genetic load will generally, when grown in the same conditions, have more surviving offspring than the average individual from a population with a high genetic load. Genetic load can also be seen as reduced fitness at the population level compared to what the population would have if all individuals had the reference high-fitness genotype. High genetic load may put a population in danger of extinction.
Mutation–selection balance is an equilibrium in the number of deleterious alleles in a population that occurs when the rate at which deleterious alleles are created by mutation equals the rate at which deleterious alleles are eliminated by selection. The majority of genetic mutations are neutral or deleterious; beneficial mutations are relatively rare. The resulting influx of deleterious mutations into a population over time is counteracted by negative selection, which acts to purge deleterious mutations. Setting aside other factors, the equilibrium number of deleterious alleles is then determined by a balance between the deleterious mutation rate and the rate at which selection purges those mutations.

Genetic distance is a measure of the genetic divergence between species or between populations within a species, whether the distance measures time from common ancestor or degree of differentiation. Populations with many similar alleles have small genetic distances. This indicates that they are closely related and have a recent common ancestor.
Coalescent theory is a model of how alleles sampled from a population may have originated from a common ancestor. In the simplest case, coalescent theory assumes no recombination, no natural selection, and no gene flow or population structure, meaning that each variant is equally likely to have been passed from one generation to the next. The model looks backward in time, merging alleles into a single ancestral copy according to a random process in coalescence events. Under this model, the expected time between successive coalescence events increases almost exponentially back in time. Variance in the model comes from both the random passing of alleles from one generation to the next, and the random occurrence of mutations in these alleles.
Genetic hitchhiking, also called genetic draft or the hitchhiking effect, is when an allele changes frequency not because it itself is under natural selection, but because it is near another gene that is undergoing a selective sweep and that is on the same DNA chain. When one gene goes through a selective sweep, any other nearby polymorphisms that are in linkage disequilibrium will tend to change their allele frequencies too. Selective sweeps happen when newly appeared mutations are advantageous and increase in frequency. Neutral or even slightly deleterious alleles that happen to be close by on the chromosome 'hitchhike' along with the sweep. In contrast, effects on a neutral locus due to linkage disequilibrium with newly appeared deleterious mutations are called background selection. Both genetic hitchhiking and background selection are stochastic (random) evolutionary forces, like genetic drift.

The Neutral Theory of Molecular Evolution is an influential monograph written in 1983 by Japanese evolutionary biologist Motoo Kimura. While the neutral theory of molecular evolution existed since his article in 1968, Kimura felt the need to write a monograph with up-to-date information and evidences showing the importance of his theory in evolution.
Neutral mutations are changes in DNA sequence that are neither beneficial nor detrimental to the ability of an organism to survive and reproduce. In population genetics, mutations in which natural selection does not affect the spread of the mutation in a species are termed neutral mutations. Neutral mutations that are inheritable and not linked to any genes under selection will be lost or will replace all other alleles of the gene. That loss or fixation of the gene proceeds based on random sampling known as genetic drift. A neutral mutation that is in linkage disequilibrium with other alleles that are under selection may proceed to loss or fixation via genetic hitchhiking and/or background selection.
Background selection describes the loss of genetic diversity at a locus due to negative selection against deleterious alleles with which it is in linkage disequilibrium. The name emphasizes the fact that the genetic background, or genomic environment, of a mutation has a significant impact on whether it will be preserved versus lost from a population. Background selection contradicts the assumption of the neutral theory of molecular evolution that the fixation or loss of a neutral allele can be described by one-locus models of genetic drift, independently from other loci. As well as reducing neutral nucleotide diversity, background selection reduces the fixation probability of beneficial mutations, and increases the fixation probability of deleterious mutations.
The nearly neutral theory of molecular evolution is a modification of the neutral theory of molecular evolution that accounts for the fact that not all mutations are either so deleterious such that they can be ignored, or else neutral. Slightly deleterious mutations are reliably purged only when their selection coefficient are greater than one divided by the effective population size. In larger populations, a higher proportion of mutations exceed this threshold for which genetic drift cannot overpower selection, leading to fewer fixation events and so slower molecular evolution.
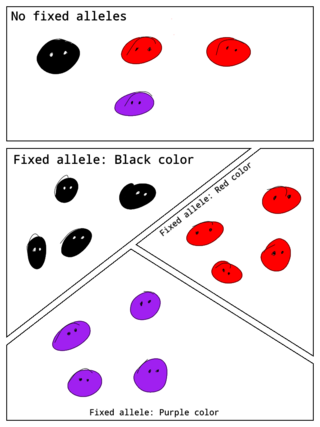
In population genetics, a fixed allele is an allele that is the only variant that exists for that gene in a population. A fixed allele is homozygous for all members of the population. The process by which alleles become fixed is called fixation.
The McDonald–Kreitman test is a statistical test often used by evolutionary and population biologists to detect and measure the amount of adaptive evolution within a species by determining whether adaptive evolution has occurred, and the proportion of substitutions that resulted from positive selection. To do this, the McDonald–Kreitman test compares the amount of variation within a species (polymorphism) to the divergence between species (substitutions) at two types of sites, neutral and nonneutral. A substitution refers to a nucleotide that is fixed within one species, but a different nucleotide is fixed within a second species at the same base pair of homologous DNA sequences. A site is nonneutral if it is either advantageous or deleterious. The two types of sites can be either synonymous or nonsynonymous within a protein-coding region. In a protein-coding sequence of DNA, a site is synonymous if a point mutation at that site would not change the amino acid, also known as a silent mutation. Because the mutation did not result in a change in the amino acid that was originally coded for by the protein-coding sequence, the phenotype, or the observable trait, of the organism is generally unchanged by the silent mutation. A site in a protein-coding sequence of DNA is nonsynonymous if a point mutation at that site results in a change in the amino acid, resulting in a change in the organism's phenotype. Typically, silent mutations in protein-coding regions are used as the "control" in the McDonald–Kreitman test.
Genetic purging is the increased pressure of natural selection against deleterious alleles prompted by inbreeding.
In population genetics, the allele frequency spectrum, sometimes called the site frequency spectrum, is the distribution of the allele frequencies of a given set of loci in a population or sample. Because an allele frequency spectrum is often a summary of or compared to sequenced samples of the whole population, it is a histogram with size depending on the number of sequenced individual chromosomes. Each entry in the frequency spectrum records the total number of loci with the corresponding derived allele frequency. Loci contributing to the frequency spectrum are assumed to be independently changing in frequency. Furthermore, loci are assumed to be biallelic, although extensions for multiallelic frequency spectra exist.




















