Related Research Articles
Aplastic anemia is a disease in which the body fails to produce blood cells in sufficient numbers. Blood cells are produced in the bone marrow by stem cells that reside there. Aplastic anaemia causes a deficiency of all blood cell types: red blood cells, white blood cells, and platelets.
Glycosylphosphatidylinositol, or glycophosphatidylinositol, or GPI in short, is a phosphoglyceride that can be attached to the C-terminus of a protein during posttranslational modification. Proteins containing a GPI anchor play key roles in a wide variety of biological processes. GPI is composed of a phosphatidylinositol group linked through a carbohydrate-containing linker and via an ethanolamine phosphate (EtNP) bridge to the C-terminal amino acid of a mature protein. The two fatty acids within the hydrophobic phosphatidyl-inositol group anchor the protein to the cell membrane.

Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, life-threatening disease of the blood characterized by destruction of red blood cells by the complement system, a part of the body's innate immune system. This destructive process occurs due to the presence of defective surface protein DAF on the red blood cell, which normally functions to inhibit such immune reactions. Since the complement cascade attacks the red blood cells within the blood vessels of the circulatory system, the red blood cell destruction (hemolysis) is considered an intravascular hemolytic anemia. Other key features of the disease, such as the high incidence of blood clot formation, are incompletely understood.

The complement system, also known as complement cascade, is a part of the immune system that enhances (complements) the ability of antibodies and phagocytic cells to clear microbes and damaged cells from an organism, promote inflammation, and attack the pathogen's cell membrane. It is part of the innate immune system, which is not adaptable and does not change during an individual's lifetime. The complement system can, however, be recruited and brought into action by antibodies generated by the adaptive immune system.
Hemolytic anemia is a form of anemia due to hemolysis, the abnormal breakdown of red blood cells (RBCs), either in the blood vessels or elsewhere in the human body (extravascular). This most commonly occurs within the spleen, but also can occur in the reticuloendothelial system or mechanically. Hemolytic anemia accounts for 5% of all existing anemias. It has numerous possible consequences, ranging from general symptoms to life-threatening systemic effects. The general classification of hemolytic anemia is either intrinsic or extrinsic. Treatment depends on the type and cause of the hemolytic anemia.

Budd–Chiari syndrome is a very rare condition, affecting one in a million adults. The condition is caused by occlusion of the hepatic veins that drain the liver. It presents with the classical triad of abdominal pain, ascites, and liver enlargement. The formation of a blood clot within the hepatic veins can lead to Budd–Chiari syndrome. The syndrome can be fulminant, acute, chronic, or asymptomatic. Subacute presentation is the most common form.
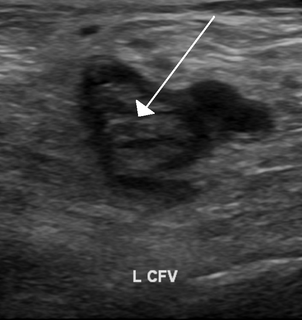
Thrombophilia is an abnormality of blood coagulation that increases the risk of thrombosis. Such abnormalities can be identified in 50% of people who have an episode of thrombosis that was not provoked by other causes. A significant proportion of the population has a detectable thrombophilic abnormality, but most of these develop thrombosis only in the presence of an additional risk factor.
Paroxysmal cold hemoglobinuria (PCH) is an autoimmune hemolytic anemia featured by complement-mediated intravascular hemolysis after cold exposure. It can present as an acute non-recurrent postinfectious event in children, or chronic relapsing episodes in adults with hematological malignancies or tertiary syphilis. Described by Julius Donath (1870–1950) and Karl Landsteiner (1868–1943) in 1904, PCH is one of the first clinical entities recognized as an autoimmune disorder.

Factor D a protein which in humans is encoded by the CFD gene. Factor D is involved in the alternative complement pathway of the complement system where it cleaves factor B.

Complement decay-accelerating factor, also known as CD55 or DAF, is a protein that, in humans, is encoded by the CD55 gene.
Eculizumab, sold under the trade name Soliris among others, is a medication used to treat paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and neuromyelitis optica. In people with PNH, it reduces both the destruction of red blood cells and need for blood transfusion, but does not appear to affect the risk of death. Eculizumab was the first drug approved for each of its uses, and its approval was granted based on small trials. It is given in a clinic by intravenous (IV) infusion.

CD59 glycoprotein, also known as MAC-inhibitory protein (MAC-IP), membrane inhibitor of reactive lysis (MIRL), or protectin, is a protein that in humans is encoded by the CD59 gene. It belongs to the LY6/uPAR/alpha-neurotoxin protein family.

Phosphatidylinositol N-acetylglucosaminyltransferase subunit A is the catalytic subunit of the phosphatidylinositol N-acetylglucosaminyltransferase enzyme, which in humans is encoded by the PIGA gene.

Phosphatidylinositol N-acetylglucosaminyltransferase subunit H is an enzyme that in humans is encoded by the PIGH gene. The PIGH gene is located on the reverse strand of chromosome 14 in humans, and is neighbored by TMEM229B.
The Ham test is a blood test used in the diagnosis of paroxysmal nocturnal hemoglobinuria (PNH). Patient red blood cells (RBCs) are placed in mild acid; a positive result indicates PNH or congenital dyserythropoietic anemia. This is now an obsolete test for diagnosing PNH due to its low sensitivity and specificity.
CD55deficiency, also called DAF deficiency or CHAPLE syndrome, is a rare disease characterized by complement-mediated autoimmune hemolysis and paroxysmal nocturnal hemoglobinuria. The protein CD55 helps to regulate the complement cascade, part of the innate immune system, by regulating the amplification phase. When CD55 is absent, the complement system attacks red blood cells and causes them to be destroyed (hemolysis).
Ravulizumab, sold under the brand name Ultomiris, is a humanized monoclonal antibody complement inhibitor medication designed for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. It is designed to bind to and prevent the activation of Complement component 5 (C5).
Donath–Landsteiner hemolytic anemia (DLHA) is a result of cold-reacting antibody immunoglobulin (Ig) induced hemolytic response inside vessels leading to anemia and, thus, a cold antibody autoimmune hemolytic anemias (CAAHA).
Cold sensitive antibodies (CSA) are antibodies sensitive to cold temperature. Some cold sensitive antibodies are pathological and can lead to blood disorder. These pathological cold sensitive antibodies include cold agglutinins, Donath-Landsteniner antibodies, and cryoglobulins which are the culprits of cold agglutinin disease, paroxysmal cold hemoglobinuria in the process of Donath-Landsteiner hemolytic anemia, and vasculitis, respectively.
Pig-a gene mutation assay is a flow cytometry-based method for detecting mammalian cells that have inactivating mutations in the endogenous X-linked reporter gene called phosphatidyl inositolglycan calss A gene. PIG-A is involved in the synthesis of glycosylphosphatidylinositol (GPI), an anchor molecule that tethers multiple protein marker molecules at the surface of the cells. When the sample containing wild-type and PIG-A mutant cells is labeled with fluorescent antibodies raised against GPI-anchored protein markers the wild-type cells will fluoresce and PIG-A mutant cells will not. The fraction of non-fluorescent PIG-A mutant cells in the antibody-labeled sample can be efficiently determined on any of the modern high throughput flow cytometers. The PIG-A mutant frequency can be determined with high accuracy within minutes by processing samples containing a total of a million cells or more.
References
- 1 2 Brodsky, RA (2009). "How I treat paroxysmal nocturnal hemoglobinuria". Blood. 113 (26): 6522–7. doi:10.1182/blood-2009-03-195966. PMC 2710914 . PMID 19372253.
- ↑ Parker C, Omine M, Richards S, et al. (2005). "Diagnosis and management of paroxysmal nocturnal hemoglobinuria". Blood. 106 (12): 3699–709. doi:10.1182/blood-2005-04-1717. PMC 1895106 . PMID 16051736.