
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
In computational linguistics and computer science, edit distance is a string metric, i.e. a way of quantifying how dissimilar two strings are to one another, that is measured by counting the minimum number of operations required to transform one string into the other. Edit distances find applications in natural language processing, where automatic spelling correction can determine candidate corrections for a misspelled word by selecting words from a dictionary that have a low distance to the word in question. In bioinformatics, it can be used to quantify the similarity of DNA sequences, which can be viewed as strings of the letters A, C, G and T.

In computer science, a suffix tree is a compressed trie containing all the suffixes of the given text as their keys and positions in the text as their values. Suffix trees allow particularly fast implementations of many important string operations.

In computer science, approximate string matching is the technique of finding strings that match a pattern approximately. The problem of approximate string matching is typically divided into two sub-problems: finding approximate substring matches inside a given string and finding dictionary strings that match the pattern approximately.
In computational phylogenetics, tree alignment is a computational problem concerned with producing multiple sequence alignments, or alignments of three or more sequences of DNA, RNA, or protein. Sequences are arranged into a phylogenetic tree, modeling the evolutionary relationships between species or taxa. The edit distances between sequences are calculated for each of the tree's internal vertices, such that the sum of all edit distances within the tree is minimized. Tree alignment can be accomplished using one of several algorithms with various trade-offs between manageable tree size and computational effort.
Sequential pattern mining is a topic of data mining concerned with finding statistically relevant patterns between data examples where the values are delivered in a sequence. It is usually presumed that the values are discrete, and thus time series mining is closely related, but usually considered a different activity. Sequential pattern mining is a special case of structured data mining.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.

Eugene Wimberly "Gene" Myers, Jr. is an American computer scientist and bioinformatician, who is best known for contributing to the early development of the NCBI's BLAST tool for sequence analysis.
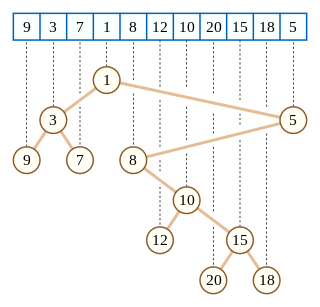
In computer science, a Cartesian tree is a binary tree derived from a sequence of distinct numbers. To construct the Cartesian tree, set its root to be the minimum number in the sequence, and recursively construct its left and right subtrees from the subsequences before and after this number. It is uniquely defined as a min-heap whose symmetric (in-order) traversal returns the original sequence.
Uzi Vishkin is a computer scientist at the University of Maryland, College Park, where he is Professor of Electrical and Computer Engineering at the University of Maryland Institute for Advanced Computer Studies (UMIACS). Uzi Vishkin is known for his work in the field of parallel computing. In 1996, he was inducted as a Fellow of the Association for Computing Machinery, with the following citation: "One of the pioneers of parallel algorithms research, Dr. Vishkin's seminal contributions played a leading role in forming and shaping what thinking in parallel has come to mean in the fundamental theory of Computer Science."
Explicit Multi-Threading (XMT) is a computer science paradigm for building and programming parallel computers designed around the parallel random-access machine (PRAM) parallel computational model. A more direct explanation of XMT starts with the rudimentary abstraction that made serial computing simple: that any single instruction available for execution in a serial program executes immediately. A consequence of this abstraction is a step-by-step (inductive) explication of the instruction available next for execution. The rudimentary parallel abstraction behind XMT, dubbed Immediate Concurrent Execution (ICE) in Vishkin (2011), is that indefinitely many instructions available for concurrent execution execute immediately. A consequence of ICE is a step-by-step (inductive) explication of the instructions available next for concurrent execution. Moving beyond the serial von Neumann computer, the aspiration of XMT is that computer science will again be able to augment mathematical induction with a simple one-line computing abstraction.
In computer science, a compressed suffix array is a compressed data structure for pattern matching. Compressed suffix arrays are a general class of data structure that improve on the suffix array. These data structures enable quick search for an arbitrary string with a comparatively small index.

Phylo is an experimental video game about multiple sequence alignment optimisation. Developed by the McGill Centre for Bioinformatics, it was originally released as a free Flash game in November 2010. Designed as a game with a purpose, players solve pattern-matching puzzles that represent nucleotide sequences of different phylogenetic taxa to optimize alignments over a computer algorithm. By aligning together each nucleotide sequence, represented as differently coloured blocks, players attempt to create the highest point value score for each set of sequences by matching as many colours as possible and minimizing gaps.
In combinatorial mathematics and theoretical computer science, heavy path decomposition is a technique for decomposing a rooted tree into a set of paths. In a heavy path decomposition, each non-leaf node selects one "heavy edge", the edge to the child that has the greatest number of descendants. The selected edges form the paths of the decomposition.
In bioinformatics, alignment-free sequence analysis approaches to molecular sequence and structure data provide alternatives over alignment-based approaches.

Ron Shamir is an Israeli professor of computer science known for his work in graph theory and in computational biology. He holds the Raymond and Beverly Sackler Chair in Bioinformatics, and is the founder and former head of the Edmond J. Safra Center for Bioinformatics at Tel Aviv University.
In mathematics and computer science, graph edit distance (GED) is a measure of similarity between two graphs. The concept of graph edit distance was first formalized mathematically by Alberto Sanfeliu and King-Sun Fu in 1983. A major application of graph edit distance is in inexact graph matching, such as error-tolerant pattern recognition in machine learning.
Daniel Mier Gusfield is an American computer scientist, Distinguished Professor of Computer Science at the University of California, Davis. Gusfield is known for his research in combinatorial optimization and computational biology.

Srinivas Aluru is a professor in the School of Computational Science and Engineering at Georgia Institute of Technology, and co-Executive Director for the Georgia Tech Interdisciplinary Research Institute in Data Engineering and Science. His main areas of research are high performance computing, data science, bioinformatics and systems biology, combinatorial methods in scientific computing, and string algorithms. Aluru is a Fellow of the American Association for the Advancement of Science (AAAS) and the Institute for Electrical and Electronic Engineers (IEEE). He is best known for his research contributions in parallel algorithms and applications, interdisciplinary research in bioinformatics and computational biology, and particularly the intersection of these two fields.
In bioinformatics, a spaced seed is a pattern of relevant and irrelevant positions in a biosequence and a method of approximate string matching that allows for substitutions. They are a straightforward modification to the earliest heuristic-based alignment efforts that allow for minor differences between the sequences of interest. Spaced seeds have been used in homology search., alignment, assembly, and metagenomics. They are usually represented as a sequence of zeroes and ones, where a one indicates relevance and a zero indicates irrelevance at the given position. Some visual representations use pound signs for relevant and dashes or asterisks for irrelevant positions.
