
In genetics, complementary DNA (cDNA) is DNA synthesized from a single-stranded RNA template in a reaction catalyzed by the enzyme reverse transcriptase. cDNA is often used to clone eukaryotic genes in prokaryotes. When scientists want to express a specific protein in a cell that does not normally express that protein, they will transfer the cDNA that codes for the protein to the recipient cell. In molecular biology, cDNA is also generated to analyze transcriptomic profiles in bulk tissue, single cells, or single nuclei in assays such as microarrays and RNA-seq.
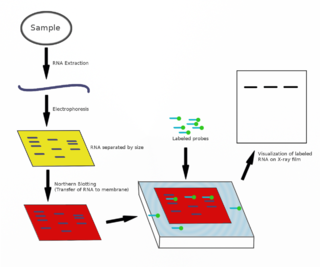
The northern blot, or RNA blot, is a technique used in molecular biology research to study gene expression by detection of RNA in a sample.

Gene expression is the process by which information from a gene is used in the synthesis of a functional gene product that enables it to produce end products, protein or non-coding RNA, and ultimately affect a phenotype, as the final effect. These products are often proteins, but in non-protein-coding genes such as transfer RNA (tRNA) and small nuclear RNA (snRNA), the product is a functional non-coding RNA. Gene expression is summarized in the central dogma of molecular biology first formulated by Francis Crick in 1958, further developed in his 1970 article, and expanded by the subsequent discoveries of reverse transcription and RNA replication.

A DNA microarray is a collection of microscopic DNA spots attached to a solid surface. Scientists use DNA microarrays to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome. Each DNA spot contains picomoles of a specific DNA sequence, known as probes. These can be a short section of a gene or other DNA element that are used to hybridize a cDNA or cRNA sample under high-stringency conditions. Probe-target hybridization is usually detected and quantified by detection of fluorophore-, silver-, or chemiluminescence-labeled targets to determine relative abundance of nucleic acid sequences in the target. The original nucleic acid arrays were macro arrays approximately 9 cm × 12 cm and the first computerized image based analysis was published in 1981. It was invented by Patrick O. Brown. An example of its application is in SNPs arrays for polymorphisms in cardiovascular diseases, cancer, pathogens and GWAS analysis. It is also used for the identification of structural variations and the measurement of gene expression.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "gene-by-gene" approach.
The transcriptome is the set of all RNA transcripts, including coding and non-coding, in an individual or a population of cells. The term can also sometimes be used to refer to all RNAs, or just mRNA, depending on the particular experiment. The term transcriptome is a portmanteau of the words transcript and genome; it is associated with the process of transcript production during the biological process of transcription.
Rapid amplification of cDNA ends (RACE) is a technique used in molecular biology to obtain the full length sequence of an RNA transcript found within a cell. RACE results in the production of a cDNA copy of the RNA sequence of interest, produced through reverse transcription, followed by PCR amplification of the cDNA copies. The amplified cDNA copies are then sequenced and, if long enough, should map to a unique genomic region. RACE is commonly followed up by cloning before sequencing of what was originally individual RNA molecules. A more high-throughput alternative which is useful for identification of novel transcript structures, is to sequence the RACE-products by next generation sequencing technologies.
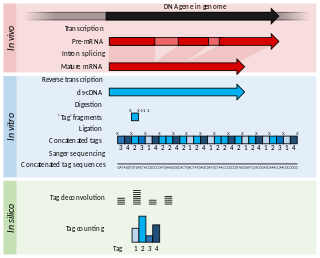
Serial Analysis of Gene Expression (SAGE) is a transcriptomic technique used by molecular biologists to produce a snapshot of the messenger RNA population in a sample of interest in the form of small tags that correspond to fragments of those transcripts. Several variants have been developed since, most notably a more robust version, LongSAGE, RL-SAGE and the most recent SuperSAGE. Many of these have improved the technique with the capture of longer tags, enabling more confident identification of a source gene.
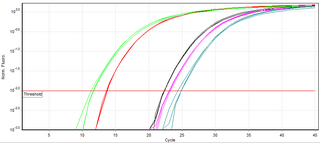
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.

Cytochrome P450 2F1 is a protein that in humans is encoded by the CYP2F1 gene.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
Trans-Spliced Exon Coupled RNA End Determination (TEC-RED) is a transcriptomic technique that, like SAGE, allows for the digital detection of messenger RNA sequences. Unlike SAGE, detection and purification of transcripts from the 5’ end of the messenger RNA require the presence of a trans-spliced leader sequence.

Long non-coding RNAs are a type of RNA, generally defined as transcripts more than 200 nucleotides that are not translated into protein. This arbitrary limit distinguishes long ncRNAs from small non-coding RNAs, such as microRNAs (miRNAs), small interfering RNAs (siRNAs), Piwi-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), and other short RNAs. Long intervening/intergenic noncoding RNAs (lincRNAs) are sequences of lncRNA which do not overlap protein-coding genes.
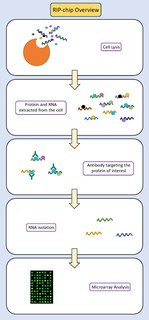
RIP-chip is a molecular biology technique which combines RNA immunoprecipitation with a microarray. The purpose of this technique is to identify which RNA sequences interact with a particular RNA binding protein of interest in vivo. It can also be used to determine relative levels of gene expression, to identify subsets of RNAs which may be co-regulated, or to identify RNAs that may have related functions. This technique provides insight into the post-transcriptional gene regulation which occurs between RNA and RNA binding proteins.

RNA-Seq is a sequencing technique which uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment, analyzing the continuously changing cellular transcriptome.
The reverse northern blot is a method by which gene expression patterns may be analyzed by comparing isolated RNA molecules from a tester sample to samples in a control cDNA library. It is a variant of the northern blot in which the nucleic acid immobilized on a membrane is a collection of isolated DNA fragments rather than RNA, and the probe is RNA extracted from a tissue and radioactively labelled. A reverse northern blot can be used to profile expression levels of particular sets of RNA sequences in a tissue or to determine presence of a particular RNA sequence in a sample. Although DNA Microarrays and newer next-generation techniques have generally supplanted reverse northern blotting, it is still utilized today and provides a relatively cheap and easy means of defining expression of large sets of genes.
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
Single-cell sequencing examines the sequence information from individual cells with optimized next-generation sequencing technologies, providing a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. For example, in cancer, sequencing the DNA of individual cells can give information about mutations carried by small populations of cells. In development, sequencing the RNAs expressed by individual cells can give insight into the existence and behavior of different cell types. In microbial systems, a population of the same species can appear genetically clonal. Still, single-cell sequencing of RNA or epigenetic modifications can reveal cell-to-cell variability that may help populations rapidly adapt to survive in changing environments.
Single-cell transcriptomics examines the gene expression level of individual cells in a given population by simultaneously measuring the RNA concentration of hundreds to thousands of genes. Single-cell transcriptomics makes it possible to unravel heterogeneous cell populations, reconstruct cellular developmental pathways, and model transcriptional dynamics — all previously masked in bulk RNA sequencing.
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst non-coding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. Transcriptomics technologies provide a broad account of which cellular processes are active and which are dormant. A major challenge in molecular biology is to understand how a single genome gives rise to a variety of cells. Another is how gene expression is regulated.
