Related Research Articles

Chromosomal crossover, or crossing over, is the exchange of genetic material during sexual reproduction between two homologous chromosomes' non-sister chromatids that results in recombinant chromosomes. It is one of the final phases of genetic recombination, which occurs in the pachytene stage of prophase I of meiosis during a process called synapsis. Synapsis begins before the synaptonemal complex develops and is not completed until near the end of prophase I. Crossover usually occurs when matching regions on matching chromosomes break and then reconnect to the other chromosome.

Genetic recombination is the exchange of genetic material between different organisms which leads to production of offspring with combinations of traits that differ from those found in either parent. In eukaryotes, genetic recombination during meiosis can lead to a novel set of genetic information that can be further passed on from parents to offspring. Most recombination occurs naturally and can be classified into two types: (1) interchromosomal recombination, occurring through independent assortment of alleles whose loci are on different but homologous chromosomes ; & (2) intrachromosomal recombination, occurring through crossing over.
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.
A genetic screen or mutagenesis screen is an experimental technique used to identify and select individuals who possess a phenotype of interest in a mutagenized population. Hence a genetic screen is a type of phenotypic screen. Genetic screens can provide important information on gene function as well as the molecular events that underlie a biological process or pathway. While genome projects have identified an extensive inventory of genes in many different organisms, genetic screens can provide valuable insight as to how those genes function.
A quantitative trait locus (QTL) is a locus that correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.
In population genetics, linkage disequilibrium (LD) is the non-random association of alleles at different loci in a given population. Loci are said to be in linkage disequilibrium when the frequency of association of their different alleles is higher or lower than expected if the loci were independent and associated randomly.

Comparative genomics is a field of biological research in which the genomic features of different organisms are compared. The genomic features may include the DNA sequence, genes, gene order, regulatory sequences, and other genomic structural landmarks. In this branch of genomics, whole or large parts of genomes resulting from genome projects are compared to study basic biological similarities and differences as well as evolutionary relationships between organisms. The major principle of comparative genomics is that common features of two organisms will often be encoded within the DNA that is evolutionarily conserved between them. Therefore, comparative genomic approaches start with making some form of alignment of genome sequences and looking for orthologous sequences in the aligned genomes and checking to what extent those sequences are conserved. Based on these, genome and molecular evolution are inferred and this may in turn be put in the context of, for example, phenotypic evolution or population genetics.

A DNA segment is identical by state (IBS) in two or more individuals if they have identical nucleotide sequences in this segment. An IBS segment is identical by descent (IBD) in two or more individuals if they have inherited it from a common ancestor without recombination, that is, the segment has the same ancestral origin in these individuals. DNA segments that are IBD are IBS per definition, but segments that are not IBD can still be IBS due to the same mutations in different individuals or recombinations that do not alter the segment.
Gene conversion is the process by which one DNA sequence replaces a homologous sequence such that the sequences become identical after the conversion event. Gene conversion can be either allelic, meaning that one allele of the same gene replaces another allele, or ectopic, meaning that one paralogous DNA sequence converts another.
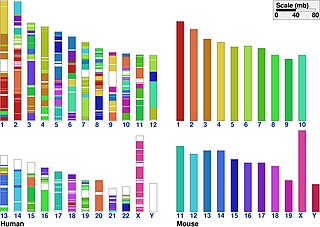
In genetics, the term synteny refers to two related concepts:
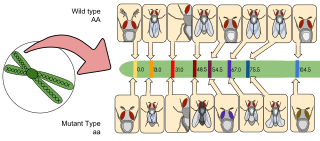
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.

Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals. Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs. Approximately two-thirds of the entire human genome may be composed of repeats and 4.8–9.5% of the human genome can be classified as copy number variations. In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.
Coalescent theory is a model of how alleles sampled from a population may have originated from a common ancestor. In the simplest case, coalescent theory assumes no recombination, no natural selection, and no gene flow or population structure, meaning that each variant is equally likely to have been passed from one generation to the next. The model looks backward in time, merging alleles into a single ancestral copy according to a random process in coalescence events. Under this model, the expected time between successive coalescence events increases almost exponentially back in time. Variance in the model comes from both the random passing of alleles from one generation to the next, and the random occurrence of mutations in these alleles.
Recombination hotspots are regions in a genome that exhibit elevated rates of recombination relative to a neutral expectation. The recombination rate within hotspots can be hundreds of times that of the surrounding region. Recombination hotspots result from higher DNA break formation in these regions, and apply to both mitotic and meiotic cells. This appellation can refer to recombination events resulting from the uneven distribution of programmed meiotic double-strand breaks.
In genetics, completelinkage is defined as the state in which two loci are so close together that alleles of these loci are virtually never separated by crossing over. The closer the physical location of two genes on the DNA, the less likely they are to be separated by a crossing-over event. In the case of male Drosophila there is complete absence of recombinant types due to absence of crossing over. This means that all of the genes that start out on a single chromosome, will end up on that same chromosome in their original configuration. In the absence of recombination, only parental phenotypes are expected.
Population genomics is the large-scale comparison of DNA sequences of populations. Population genomics is a neologism that is associated with population genetics. Population genomics studies genome-wide effects to improve our understanding of microevolution so that we may learn the phylogenetic history and demography of a population.
In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
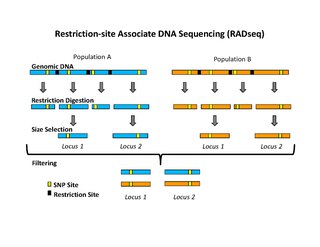
Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome. Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs). Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.
A recombinant inbred strain or recombinant inbred line (RIL) is an organism with chromosomes that incorporate an essentially permanent set of recombination events between chromosomes inherited from two or more inbred strains. F1 and F2 generations are produced by intercrossing the inbred strains; pairs of the F2 progeny are then mated to establish inbred strains through long-term inbreeding.
A sequence related amplified polymorphism (SRAP) is a molecular technique, developed by G. Li and C. F. Quiros in 2001, for detecting genetic variation in the open reading frames (ORFs) of genomes of plants and related organisms.
References
- ↑ Dear PH, Cook PR (September 1989). "Happy mapping: a proposal for linkage mapping the human genome". Nucleic Acids Res. 17 (17): 6795–807. doi:10.1093/nar/17.17.6795. PMC 318413 . PMID 2780310.
- ↑ Piper MB, Bankier AT, Dear PH (December 1998). "A HAPPY map of Cryptosporidium parvum". Genome Res. 8 (12): 1299–307. doi:10.1101/gr.8.12.1299. PMC 310802 . PMID 9872984.
- ↑ Hamilton EP, Dear PH, Rowland T, Saks K, Eisen JA, Orias E (October 2006). "Use of HAPPY mapping for the higher order assembly of the Tetrahymena genome". Genomics. 88 (4): 443–51. doi:10.1016/j.ygeno.2006.05.002. PMC 3169840 . PMID 16782302.
- ↑ McCaughan F, Darai-Ramqvist E, Bankier AT, Konfortov BA, Foster N, George PJ, Rabbitts TH, Kost-Alimova M, Rabbitts PH, Dear PH (November 2008). "Microdissection molecular copy-number counting (microMCC)--unlocking cancer archives with digital PCR". J. Pathol. 216 (3): 307–16. doi: 10.1002/path.2413 . PMID 18773450.