Hemoglobin or haemoglobin, abbreviated Hb or Hgb, is the iron-containing oxygen-transport metalloprotein in the red blood cells (erythrocytes) of almost all vertebrates as well as the tissues of some invertebrates. Hemoglobin in blood carries oxygen from the lungs or gills to the rest of the body. There it releases the oxygen to permit aerobic respiration to provide energy to power the functions of the organism in the process called metabolism. A healthy individual has 12 to 20 grams of hemoglobin in every 100 mL of blood.

Haptoglobin is the protein that in humans is encoded by the HP gene. In blood plasma, haptoglobin binds to free hemoglobin, compared to hemopexin that binds to free heme, released from erythrocytes with high affinity, and thereby inhibits its deleterious oxidative activity. The haptoglobin-hemoglobin complex will then be removed by the reticuloendothelial system.

Thalassemias are inherited blood disorders characterized by decreased hemoglobin production. Symptoms depend on the type and can vary from none to severe. Often there is mild to severe anemia. Anemia can result in feeling tired and pale skin. There may also be bone problems, an enlarged spleen, yellowish skin, and dark urine. Slow growth may occur in children.
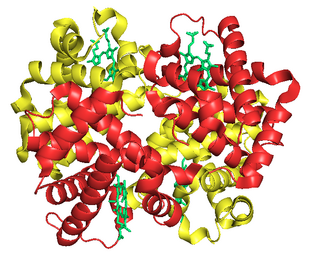
Fetal hemoglobin, or foetal haemoglobin is the main oxygen carrier protein in the human fetus. Hemoglobin F is found in fetal red blood cells, and is involved in transporting oxygen from the mother's bloodstream to organs and tissues in the fetus. It is produced at around 6 weeks of pregnancy and the levels remain high after birth until the baby is roughly 2–4 months old. Hemoglobin F has a different composition from the adult forms of hemoglobin, which allows it to bind oxygen more strongly. This way, the developing fetus is able to retrieve oxygen from the mother's bloodstream, which occurs through the placenta found in the mother's uterus.

The crocodile icefish or white-blooded fish comprise a family (Channichthyidae) of notothenioid fishes found in the Southern Ocean around Antarctica. They are the only known vertebrates to lack hemoglobin in their blood as adults. Icefish populations are known to reside in the Atlantic and Indian sectors of the Southern Ocean, as well as the continental shelf waters surrounding Antarctica. Water temperatures in these regions remain relatively stable, generally ranging from −1.8 to 2 °C. One icefish, Champsocephalus esox, is distributed north of the Antarctic Polar Frontal Zone. At least 16 species of crocodile icefish are currently recognized, although eight additional species have been proposed for the icefish genus Channichthys.
Hemoglobin A (HbA), also known as adult hemoglobin, hemoglobin A1 or α2β2, is the most common human hemoglobin tetramer, accounting for over 97% of the total red blood cell hemoglobin. Hemoglobin is an oxygen-binding protein, found in erythrocytes, which transports oxygen from the lungs to the tissues. Hemoglobin A is the most common adult form of hemoglobin and exists as a tetramer containing two alpha subunits and two beta subunits (α2β2). Hemaglobin A2 (HbA2) is a less common adult form of hemoglobin and is composed of two alpha and two delta-globin subunits. This hemoglobin makes up 1-3% of hemoglobin in adults.
Hemoglobin C is an abnormal hemoglobin in which glutamic acid residue at the 6th position of the β-globin chain is replaced with a lysine residue due to a point mutation in the HBB gene. People with one copy of the gene for hemoglobin C do not experience symptoms, but can pass the abnormal gene on to their children. Those with two copies of the gene are said to have hemoglobin C disease and can experience mild anemia. It is possible for a person to have both the gene for hemoglobin S and the gene for hemoglobin C; this state is called hemoglobin SC disease, and is generally more severe than hemoglobin C disease, but milder than sickle cell anemia.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.

Sickle cell trait describes a condition in which a person has one abnormal allele of the hemoglobin beta gene, but does not display the severe symptoms of sickle cell disease that occur in a person who has two copies of that allele. Those who are heterozygous for the sickle cell allele produce both normal and abnormal hemoglobin.
Hemoglobin Barts, abbreviated Hb Barts, is an abnormal type of hemoglobin that consists of four gamma globins. It is moderately insoluble, and therefore accumulates in the red blood cells. Hb Barts has an extremely high affinity for oxygen, so it cannot release oxygen to the tissue. Therefore, this makes it an inefficient oxygen carrier. As an embryo develops, it begins to produce alpha-globins at weeks 5–6 of development. When both of the HBA1 and HBA2 genes which code for alpha globins becomes dysfunctional, the affected fetuses will have difficulty in synthesizing a functional hemoglobin. As a result, gamma chains will accumulate and form four gamma globins. These gamma globins bind to form hemoglobin Barts. It is produced in the disease alpha-thalassemia and in the most severe of cases, it is the only form of hemoglobin in circulation. In this situation, a fetus will develop hydrops fetalis and normally die before or shortly after birth, unless intrauterine blood transfusion is performed.

Alpha-catenin functions as the primary protein link between cadherins and the actin cytoskeleton. It has been reported that the actin binding proteins vinculin and alpha-actinin can bind to alpha-catenin. It has been suggested that alpha-catenin does not bind with high affinity to both actin filaments and the E-cadherin-beta-catenin complex at the same time. It has been observed that when alpha-catenin is not in a molecular complex with beta-catenin, it dimerizes and functions to regulate actin filament assembly, possibly by competing with Arp2/3 protein. Alpha catenin exhibits significant protein dynamics.

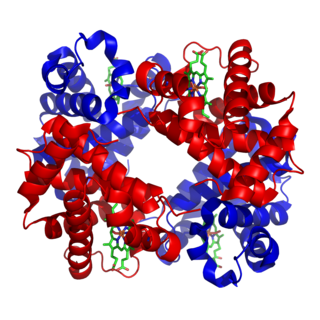
Hemoglobin subunit alpha, Hemoglobin, alpha 1, also known as HBA1, is a hemoglobin protein that in humans is encoded by the HBA1 gene.
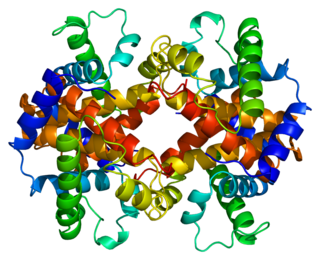
Hemoglobin subunit epsilon is a protein that in humans is encoded by the HBE1 gene.

Hemoglobin subunit zeta is a protein that in humans is encoded by the HBZ gene.
"Sickle Cell Anemia, a Molecular Disease" is a 1949 scientific paper by Linus Pauling, Harvey A. Itano, Seymour J. Singer and Ibert C. Wells that established sickle-cell anemia as a genetic disease in which affected individuals have a different form of the metalloprotein hemoglobin in their blood. The paper, published in the November 25, 1949 issue of Science, reports a difference in electrophoretic mobility between hemoglobin from healthy individuals and those with sickle-cell anemia, with those with sickle cell trait having a mixture of the two types. The paper suggests that the difference in electrophoretic mobility is probably due to a different number of ionizable amino acid residues in the protein portion of hemoglobin, and that this change in molecular structure is responsible for the sickling process. It also reports the genetic basis for the disease, consistent with the simultaneous genealogical study by James V. Neel: those with sickle-cell anemia are homozygous for the disease gene, while heterozygous individuals exhibit the usually asymptomatic condition of sickle cell trait.

Hemoglobin, alpha 2 also known as HBA2 is a gene that in humans codes for the alpha globin chain of hemoglobin.

The aldo-keto reductase family is a family of proteins that are subdivided into 16 categories; these include a number of related monomeric NADPH-dependent oxidoreductases, such as aldehyde reductase, aldose reductase, prostaglandin F synthase, xylose reductase, rho crystallin, and many others.

Hemoglobin Hopkins-2 is a mutation of the protein hemoglobin, which is responsible for the transportation of oxygen through the blood from the lungs to the musculature of the body in vertebrates. The specific mutation in Hemoglobin Hopkins-2 results in two abnormal α chains. The mutation is the result of histidine 112 being replaced with aspartic acid in the protein's polypeptide sequence. Additionally, within one of the mutated alpha chains, there are substitutes at 114 and 118, two points on the amino acid chain. This mutation can cause sickle cell anemia.
Hemoglobin O (HbO) is a rare type of hemoglobin in which there is a substitution of glutamic acid by lysine as in hemoglobin C, but at different positions. Since the amino acid substitution can occur at different positions of the β-globin chain of the protein, there are several variants. In hemoglobin O-Arab (HbO-Arab) substitution occurs at position 121, while in hemoglobin O-Padova (HbO-Padova) it is at 11 position, and in hemoglobin O Indonesia (HbOIna) it is at 116.