Related Research Articles

Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).

Spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of rare debilitating disorders characterised by the degeneration of lower motor neurons and subsequent atrophy (wasting) of various muscle groups in the body. While some SMAs lead to early infant death, other diseases of this group permit normal adult life with only mild weakness.
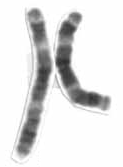
Chromosome 3 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 3 spans more than 198 million base pairs and represents about 6.5 percent of the total DNA in cells.
Dejerine–Sottas disease, also known as, Dejerine–Sottas syndrome, hereditary motor and sensory polyneuropathy type III, and Charcot–Marie–Tooth disease type 3, is a hereditary neurological disorder characterized by damage to the peripheral nerves, demyelination, and resulting progressive muscle wasting and somatosensory loss. The condition is caused by mutations in various genes and currently has no known cure.

Familial amyloid polyneuropathy, also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis abbreviated also as ATTR, or Corino de Andrade's disease, is an autosomal dominant neurodegenerative disease. It is a form of amyloidosis, and was first identified and described by Portuguese neurologist Mário Corino da Costa Andrade, in 1952. FAP is distinct from senile systemic amyloidosis (SSA), which is not inherited, and which was determined to be the primary cause of death for 70% of supercentenarians who have been autopsied. FAP can be ameliorated by liver transplantation.

Myelin protein zero is a single membrane glycoprotein which in humans is encoded by the MPZ gene. P0 is a major structural component of the myelin sheath in the peripheral nervous system (PNS). Myelin protein zero is expressed by Schwann cells and accounts for over 50% of all proteins in the peripheral nervous system, making it the most common protein expressed in the PNS. Mutations in myelin protein zero can cause myelin deficiency and are associated with neuropathies like Charcot–Marie–Tooth disease and Dejerine–Sottas disease.
Ras-related protein Rab-7a is a protein that in humans is encoded by the RAB7A gene.

Heat shock protein beta-8 is a protein that in humans is encoded by the HSPB8 gene.

In enzymology, a serine C-palmitoyltransferase (EC 2.3.1.50) is an enzyme that catalyzes the chemical reaction:

Dystonin(DST), also known as bullous pemphigoid antigen 1 (BPAG1), isoforms 1/2/3/4/5/8, is a protein that in humans is encoded by the DST gene.

Serine palmitoyltransferase, long chain base subunit 1, also known as SPTLC1, is a protein which in humans is encoded by the SPTLC1 gene.

Serine palmitoyltransferase, long chain base subunit 2, also known as SPTLC2, is a protein which in humans is encoded by the SPTLC2 gene. SPTLC2 belongs to the class-II pyridoxal-phosphate-dependent aminotransferase family.

Hereditary motor and sensory neuropathies (HMSN) is a name sometimes given to a group of different neuropathies which are all characterized by their impact upon both afferent and efferent neural communication. HMSN are characterised by atypical neural development and degradation of neural tissue. The two common forms of HMSN are either hypertrophic demyelinated nerves or complete atrophy of neural tissue. Hypertrophic condition causes neural stiffness and a demyelination of nerves in the peripheral nervous system, and atrophy causes the breakdown of axons and neural cell bodies. In these disorders, a patient experiences progressive muscle atrophy and sensory neuropathy of the extremities.
Hereditary sensory and autonomic neuropathy (HSAN) or hereditary sensory neuropathy (HSN) is a condition used to describe any of the types of this disease which inhibit sensation.
Hereditary sensory neuropathy, type II also known as HSN2 is a region of a parent protein which in humans is encoded by the WNK1 gene. It is a transcript variant of the WNK1 gene that is selectively expressed in nervous system tissues, and during development. Mutations in this exon of the WNK1 gene have been identified as causative in genetic neuropathy syndromes, and in inherited pain insensitivity.

Hereditary neuropathy with liability to pressure palsy (HNPP) is a peripheral neuropathy, a condition that affects the nerves. Pressure on the nerves can cause tingling sensations, numbness, pain, weakness, muscle atrophy and even paralysis of the affected area. In normal individuals, these symptoms disappear quickly, but in sufferers of HNPP even a short period of pressure can cause the symptoms to occur. Palsies can last from minutes or days to weeks or even months.
Distal hereditary motor neuronopathies, sometimes also called distal hereditary motor neuropathies, are a genetically and clinically heterogeneous group of motor neuron diseases that result from genetic mutations in various genes and are characterized by degeneration and loss of motor neuron cells in the anterior horn of the spinal cord and subsequent muscle atrophy.
Hereditary motor and sensory neuropathy with proximal dominance (HMSN-P) is an autosomal dominant neurodegenerative disorder that is defined by extensive involuntary and spontaneous muscle contractions, asthenia, and atrophy with distal sensory involvement following. The disease starts presenting typically in the 40s and is succeeded by a slow and continuous onslaught. Muscle spasms and muscle contractions large in number are noted, especially in the earliest stages. The presentation of HMSN-P is quite similar to amyotrophic lateral sclerosis and has common neuropathological findings. Sensory loss happens as the disease progresses, but the amount of sensation lost varies from case to case. There have been other symptoms of HMSN-P reported such as urinary disturbances and a dry cough.
Hereditary sensory and autonomic neuropathy type I or hereditary sensory neuropathy type I is a group of autosomal dominant inherited neurological diseases that affect the peripheral nervous system particularly on the sensory and autonomic functions. The hallmark of the disease is the marked loss of pain and temperature sensation in the distal parts of the lower limbs. The autonomic disturbances, if present, manifest as sweating abnormalities.
Distal hereditary motor neuropathy type V is a particular type of neuropathic disorder. In general, distal hereditary motor neuropathies affect the axons of distal motor neurons and are characterized by progressive weakness and atrophy of muscles of the extremities. It is common for them to be called "spinal forms of Charcot-Marie-Tooth disease (CMT)", because the diseases are closely related in symptoms and genetic cause. The diagnostic difference in these diseases is the presence of sensory loss in the extremities. There are seven classifications of dHMNs, each defined by patterns of inheritance, age of onset, severity, and muscle groups involved. Type V is a disorder characterized by autosomal dominance, weakness of the upper limbs that is progressive and symmetrical, and atrophy of the small muscles of the hands.
References
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Entrez Gene: Hereditary sensory neuropathy, type IB" . Retrieved 2017-02-04.
| | This article on a gene on human chromosome 3 is a stub. You can help Wikipedia by expanding it. |