Related Research Articles

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.

Molecular phylogenetics is the branch of phylogeny that analyzes genetic, hereditary molecular differences, predominately in DNA sequences, to gain information on an organism's evolutionary relationships. From these analyses, it is possible to determine the processes by which diversity among species has been achieved. The result of a molecular phylogenetic analysis is expressed in a phylogenetic tree. Molecular phylogenetics is one aspect of molecular systematics, a broader term that also includes the use of molecular data in taxonomy and biogeography.

Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.
The neutral theory of molecular evolution holds that most evolutionary changes at the molecular level, and most of the variation within and between species, are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin. The neutral theory allows for the possibility that most mutations are deleterious, but holds that because these are rapidly removed by natural selection, they do not make significant contributions to variation within and between species at the molecular level. A neutral mutation is one that does not affect an organism's ability to survive and reproduce. The neutral theory assumes that most mutations that are not deleterious are neutral rather than beneficial. Because only a fraction of gametes are sampled in each generation of a species, the neutral theory suggests that a mutant allele can arise within a population and reach fixation by chance, rather than by selective advantage.
The molecular clock is a figurative term for a technique that uses the mutation rate of biomolecules to deduce the time in prehistory when two or more life forms diverged. The biomolecular data used for such calculations are usually nucleotide sequences for DNA or amino acid sequences for proteins. The benchmarks for determining the mutation rate are often fossil or archaeological dates. The molecular clock was first tested in 1962 on the hemoglobin protein variants of various animals, and is commonly used in molecular evolution to estimate times of speciation or radiation. It is sometimes called a gene clock or an evolutionary clock.
In bioinformatics and evolutionary biology, a substitution matrix describes the rate at which one character in a sequence changes to other character states over time. It is an application of a stochastic matrix. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments, where the similarity between sequences depends on their divergence time and the substitution rates as represented in the matrix.
In biology, a substitution model describes the process from which a sequence of symbols changes into another set of traits. For example, in cladistics, each position in the sequence might correspond to a property of a species which can either be present or absent. The alphabet could then consist of "0" for absence and "1" for presence. Then the sequence 00110 could mean, for example, that a species does not have feathers or lay eggs, does have fur, is warm-blooded, and cannot breathe underwater. Another sequence 11010 would mean that a species has feathers, lays eggs, does not have fur, is warm-blooded, and cannot breathe underwater. In phylogenetics, sequences are often obtained by firstly obtaining a nucleotide or protein sequence alignment, and then taking the bases or amino acids at corresponding positions in the alignment as the characters. Sequences achieved by this might look like AGCGGAGCTTA and GCCGTAGACGC.
In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.
Computational phylogenetics is the application of computational algorithms, methods, and programs to phylogenetic analyses. The goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa. For example, these techniques have been used to explore the family tree of hominid species and the relationships between specific genes shared by many types of organisms. Traditional phylogenetics relies on morphological data obtained by measuring and quantifying the phenotypic properties of representative organisms, while the more recent field of molecular phylogenetics uses nucleotide sequences encoding genes or amino acid sequences encoding proteins as the basis for classification. Many forms of molecular phylogenetics are closely related to and make extensive use of sequence alignment in constructing and refining phylogenetic trees, which are used to classify the evolutionary relationships between homologous genes represented in the genomes of divergent species. The phylogenetic trees constructed by computational methods are unlikely to perfectly reproduce the evolutionary tree that represents the historical relationships between the species being analyzed. The historical species tree may also differ from the historical tree of an individual homologous gene shared by those species.
In genetics, the Ka/Ks ratio, also known as ω or dN/dS ratio, is used to estimate the balance between neutral mutations, purifying selection and beneficial mutations acting on a set of homologous protein-coding genes. It is calculated as the ratio of the number of nonsynonymous substitutions per non-synonymous site (Ka), in a given period of time, to the number of synonymous substitutions per synonymous site (Ks), in the same period. The latter are assumed to be neutral, so that the ratio indicates the net balance between deleterious and beneficial mutations. Values of Ka/Ks significantly above 1 are unlikely to occur without at least some of the mutations being advantageous. If beneficial mutations are assumed to make little contribution, then Ks estimates the degree of evolutionary constraint.
Neutral mutations are changes in DNA sequence that are neither beneficial nor detrimental to the ability of an organism to survive and reproduce. In population genetics, mutations in which natural selection does not affect the spread of the mutation in a species are termed neutral mutations. Neutral mutations that are inheritable and not linked to any genes under selection will either be lost or will replace all other alleles of the gene. This loss or fixation of the gene proceeds based on random sampling known as genetic drift. A neutral mutation that is in linkage disequilibrium with other alleles that are under selection may proceed to loss or fixation via genetic hitchhiking and/or background selection.
Ancestral reconstruction is the extrapolation back in time from measured characteristics of individuals to their common ancestors. It is an important application of phylogenetics, the reconstruction and study of the evolutionary relationships among individuals, populations or species to their ancestors. In the context of evolutionary biology, ancestral reconstruction can be used to recover different kinds of ancestral character states of organisms that lived millions of years ago. These states include the genetic sequence, the amino acid sequence of a protein, the composition of a genome, a measurable characteristic of an organism (phenotype), and the geographic range of an ancestral population or species. This is desirable because it allows us to examine parts of phylogenetic trees corresponding to the distant past, clarifying the evolutionary history of the species in the tree. Since modern genetic sequences are essentially a variation of ancient ones, access to ancient sequences may identify other variations and organisms which could have arisen from those sequences. In addition to genetic sequences, one might attempt to track the changing of one character trait to another, such as fins turning to legs.
Human evolutionary genetics studies how one human genome differs from another human genome, the evolutionary past that gave rise to the human genome, and its current effects. Differences between genomes have anthropological, medical, historical and forensic implications and applications. Genetic data can provide important insights into human evolution.
Orphan genes are genes without detectable homologues in other lineages. Orphans are a subset of taxonomically-restricted genes (TRGs), which are unique to a specific taxonomic level. In contrast to non-orphan TRGs, orphans are usually considered unique to a very narrow taxon, generally a species.
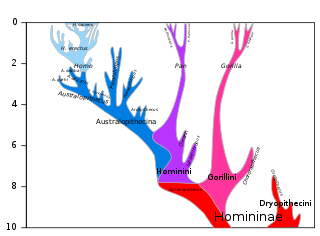
The chimpanzee–human last common ancestor (CHLCA) is the last common ancestor shared by the extant Homo (human) and Pan genera of Hominini. Due to complex hybrid speciation, it is not possible to give a precise estimate on the age of this ancestral population. While "original divergence" between populations may have occurred as early as 13 million years ago (Miocene), hybridization may have been ongoing until as recently as 4 million years ago (Pliocene).
Functional divergence is the process by which genes, after gene duplication, shift in function from an ancestral function. Functional divergence can result in either subfunctionalization, where a paralog specializes one of several ancestral functions, or neofunctionalization, where a totally new functional capability evolves. It is thought that this process of gene duplication and functional divergence is a major originator of molecular novelty and has produced the many large protein families that exist today.
Ziheng Yang FRS is a Chinese biologist. He holds the R.A. Fisher Chair of Statistical Genetics at University College London, and is the Director of R.A. Fisher Centre for Computational Biology at UCL. He was elected a Fellow of the Royal Society in 2006.
Viral phylodynamics is defined as the study of how epidemiological, immunological, and evolutionary processes act and potentially interact to shape viral phylogenies. Since the coining of the term in 2004, research on viral phylodynamics has focused on transmission dynamics in an effort to shed light on how these dynamics impact viral genetic variation. Transmission dynamics can be considered at the level of cells within an infected host, individual hosts within a population, or entire populations of hosts.

Venom in snakes and some lizards is a form of saliva that has been modified into venom over its evolutionary history. In snakes, venom has evolved to kill or subdue prey, as well as to perform other diet-related functions. The evolution of venom is thought to be responsible for the enormous expansion of snakes across the globe.
Genetic saturation is the result of multiple substitutions at the same site in a sequence, or identical substitutions in different sequences, such that the apparent sequence divergence rate is lower than the actual divergence that has occurred. When comparing two or more genetic sequences consisting of single nucleotides, differences in sequence observed are only differences in the final state of the nucleotide sequence. Single nucleotides that undergoing genetic saturation change multiple times, sometimes back to their original nucleotide or to a nucleotide common to the compared genetic sequence. Without genetic information from intermediate taxa, it is difficult to know how much, or if any saturation has occurred on an observed sequence. Genetic saturation occurs most rapidly on fast-evolving sequences, such as the hypervariable region of mitochondrial DNA, or in short tandem repeats such as on the Y-chromosome.
References
- ↑ Lopez P, Casane D, Philippe H (January 2002). "Heterotachy, an important process of protein evolution". Mol. Biol. Evol. 19 (1): 1–7. doi: 10.1093/oxfordjournals.molbev.a003973 . PMID 11752184.
- ↑ Zhong, Bojian; Oliver Deusch; Vadim V. Goremykin; David Penny; Patrick J. Biggs; Robin A. Atherton; Svetlana V. Nikiforova; Peter James Lockhart (2011). "Systematic error in seed plant phylogenomics". Genome Biology and Evolution. 3: 1340–1348. doi:10.1093/gbe/evr105. PMC 3237385 . PMID 22016337.