
Prader–Willi syndrome (PWS) is a rare genetic disorder caused by a loss of function of specific genes on chromosome 15. In newborns, symptoms include weak muscles, poor feeding, and slow development. Beginning in childhood, those affected become constantly hungry, which often leads to obesity and type 2 diabetes. Mild to moderate intellectual impairment and behavioral problems are also typical of the disorder. Often, affected individuals have a narrow forehead, small hands and feet, short height, and light skin and hair. Most are unable to have children.

Uniparental disomy (UPD) occurs when a person receives two copies of a chromosome, or of part of a chromosome, from one parent and no copy from the other. UPD can be the result of heterodisomy, in which a pair of non-identical chromosomes are inherited from one parent or isodisomy, in which a single chromosome from one parent is duplicated. Uniparental disomy may have clinical relevance for several reasons. For example, either isodisomy or heterodisomy can disrupt parent-specific genomic imprinting, resulting in imprinting disorders. Additionally, isodisomy leads to large blocks of homozygosity, which may lead to the uncovering of recessive genes, a similar phenomenon seen in inbred children of consanguineous partners.

Ubiquitin-protein ligase E3A (UBE3A) also known as E6AP ubiquitin-protein ligase (E6AP) is an enzyme that in humans is encoded by the UBE3A gene. This enzyme is involved in targeting proteins for degradation within cells.

Chromosome 15 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 15 spans about 99.7 million base pairs and represents between 3% and 3.5% of the total DNA in cells. Chromosome 15 is an acrocentric chromosome, with a very small short arm, which contains few protein coding genes among its 19 million base pairs. It has a larger long arm that is gene rich, spanning about 83 million base pairs.
The short-stature homeobox gene (SHOX), also known as short-stature-homeobox-containing gene, is a gene located on both the X and Y chromosomes, which is associated with short stature in humans if mutated or present in only one copy (haploinsufficiency).

22q13 deletion syndrome, known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

Craniofrontonasal dysplasia is a very rare X-linked malformation syndrome caused by mutations in the ephrin-B1 gene (EFNB1). Phenotypic expression varies greatly amongst affected individuals, where females are more commonly and generally more severely affected than males. Common physical malformations are: craniosynostosis of the coronal suture(s), orbital hypertelorism, bifid nasal tip, dry frizzy curled hair, longitudinal ridging and/or splitting of the nails, and facial asymmetry.
Infantile Refsum disease (IRD) is a rare autosomal recessive congenital peroxisomal biogenesis disorder within the Zellweger spectrum. These are disorders of the peroxisomes that are clinically similar to Zellweger syndrome and associated with mutations in the PEX family of genes. IRD is associated with deficient phytanic acid catabolism, as is adult Refsum disease, but they are different disorders that should not be confused.

Phosphomannomutase 2 is an enzyme that in humans is encoded by the PMM2 gene.

KCNQ1 overlapping transcript 1, also known as KCNQ1OT1, is a long non-coding RNA gene found in the KCNQ1 locus. This locus consists of 8–10 protein-coding genes, specifically expressed from the maternal allele, and the paternally expressed non-coding RNA gene KCNQ1OT1. KCNQ1OT1 and KCNQ1 are imprinted genes and are part of an imprinting control region (ICR). Mitsuya identified that KCNQ1OT1 is an antisense transcript of KCNQ1. KCNQ1OT1 is a paternally expressed allele and KCNQ1 is a maternally expressed allele. KCNQ1OT1 is a nuclear, 91 kb transcript, found in close proximity to the nucleolus in certain cell types.

18p-, also known as monosomy 18p, deletion 18p syndrome, del(18p) syndrome, partial monosomy 18p, or de Grouchy syndrome 1, is a genetic condition caused by a deletion of all or part of the short arm of chromosome 18. It occurs in about 1 of every 50,000 births.
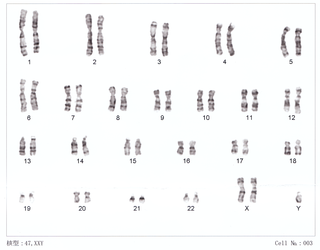
Klinefelter syndrome (KS), also known as 47,XXY, is a chromosome anomaly where a male has an extra X chromosome. These complications commonly include infertility and small, poorly functioning testicles. These symptoms are often noticed only at puberty, although this is one of the most common chromosomal disorders, occurring in one to two per 1,000 live births. It is named after American endocrinologist Harry Klinefelter, who identified the condition in the 1940s, along with his colleagues at Massachusetts General Hospital.
A Finnish heritage disease is any genetic disease or disorder that is significantly more common in people whose ancestors were ethnic Finns, natives of Finland and Northern Sweden (Meänmaa) and Northwest Russia. There are 36 rare diseases regarded as Finnish heritage diseases. The diseases are not restricted to Finns; they are genetic diseases with far wider distribution in the world, but due to founder effects and genetic isolation they are more common in Finns.

Silver–Russell syndrome, also called Silver–Russell dwarfism, is a rare congenital growth disorder. In the United States it is usually referred to as Russell–Silver syndrome, and Silver–Russell syndrome elsewhere. It is one of 200 types of dwarfism and one of five types of primordial dwarfism.
The proportions of various human Y-DNA haplogroups vary significantly from one ethnic or language group to another in Africa.

9q34 deletion syndrome is a rare genetic disorder. Terminal deletions of chromosome 9q34 have been associated with childhood hypotonia, a distinctive facial appearance and developmental disability. The facial features typically described include arched eyebrows, small head circumference, midface hypoplasia, prominent jaw and a pouting lower lip. Individuals with this disease may often have speech impediments, such as speech delays. Other characteristics of this disease include: epilepsy, congenital and urogenital defects, microcephaly, corpulence, and psychiatric disorders. From analysis of chromosomal breakpoints, as well as gene sequencing in suggestive cases, Kleefstra and colleagues identified EHMT1 as the causative gene. This gene is responsible for producing the protein histone methyltransferase which functions to alter histones. Ultimately, histone methyltransferases are important in deactivating certain genes, needed for proper growth and development. Moreover, a frameshift, missense, or nonsense error in the coding sequence of EHMT1 can result in this condition in an individual.
Autism spectrum disorder (ASD) refers to a variety of conditions typically identified by challenges with social skills, communication, speech, and repetitive sensory-motor behaviors. The 11th International Classification of Diseases (ICD-11), released in January 2021, characterizes ASD by the associated deficits in the ability to initiate and sustain two-way social communication and restricted or repetitive behavior unusual for the individual's age or situation. Although linked with early childhood, the symptoms can appear later as well. Symptoms can be detected before the age of two and experienced practitioners can give a reliable diagnosis by that age. However, official diagnosis may not occur until much older, even well into adulthood. There is a large degree of variation in how much support a person with ASD needs in day-to-day life. This can be classified by a further diagnosis of ASD level 1, level 2, or level 3. Of these, ASD level 3 describes people requiring very substantial support and who experience more severe symptoms. ASD-related deficits in nonverbal and verbal social skills can result in impediments in personal, family, social, educational, and occupational situations. This disorder tends to have a strong correlation with genetics along with other factors. More research is identifying ways in which epigenetics is linked to autism. Epigenetics generally refers to the ways in which chromatin structure is altered to affect gene expression. Mechanisms such as cytosine regulation and post-translational modifications of histones. Of the 215 genes contributing, to some extent in ASD, 42 have been found to be involved in epigenetic modification of gene expression. Some examples of ASD signs are specific or repeated behaviors, enhanced sensitivity to materials, being upset by changes in routine, appearing to show reduced interest in others, avoiding eye contact and limitations in social situations, as well as verbal communication. When social interaction becomes more important, some whose condition might have been overlooked suffer social and other exclusion and are more likely to have coexisting mental and physical conditions. Long-term problems include difficulties in daily living such as managing schedules, hypersensitivities, initiating and sustaining relationships, and maintaining jobs.
Genetic studies on Arabs refers to the analyses of the genetics of ethnic Arab people in the Middle East and North Africa. Arabs are genetically diverse as a result of their intermarriage and mixing with indigenous people of the pre-Islamic Middle East and North Africa following the Arab and Islamic expansion. Genetic ancestry components related to the Arabian Peninsula display an increasing frequency pattern from west to east over North Africa. A similar frequency pattern exist across northeastern Africa with decreasing genetic affinities to groups of the Arabian Peninsula along the Nile river valley across Sudan and the more they go south. This genetic cline of admixture is dated to the time of Arab migrations to the Maghreb and northeast Africa.
Birk-Barel syndrome is a rare genetic disorder associated with the KCNK9 gene. Signs and symptoms include intellectual disability, hypotonia, hyperactivity, and syndromic facies.

Structural Maintenance of Chromosomes flexible Hinge Domain Containing 1 (SMCHD1) is a protein that in humans is encoded by the SMCHD1 gene. Mutations in SMCHD1 are causative for development of facioscapulohumeral muscular dystrophy type 2 (FSHD2) and Bosma arhinia microphthalmia syndrome (BAMS).
