Related Research Articles

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications, the elements are more often referred to as points, nodes or vertices.
In computer graphics, level of detail (LOD) refers to the complexity of a 3D model representation. LOD can be decreased as the model moves away from the viewer or according to other metrics such as object importance, viewpoint-relative speed or position. LOD techniques increase the efficiency of rendering by decreasing the workload on graphics pipeline stages, usually vertex transformations. The reduced visual quality of the model is often unnoticed because of the small effect on object appearance when distant or moving fast.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.
OpenEye Scientific Software is an American software company founded by Anthony Nicholls in 1997. It develops large-scale molecular modelling applications and toolkits. Following OpenEye's acquisition by Cadence Design Systems for $500 million in September 2022, the company was rebranded to OpenEye Cadence Molecular Sciences and operates as a business unit under Cadence.

In the context of chemistry, molecular physics and physical chemistry and molecular modelling, a force field is a computational model that is used to describe the forces between atoms within molecules or between molecules as well as in crystals. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of the atomistic level. Force fields are usually used in molecular dynamics or Monte Carlo simulations. The parameters for a chosen energy function may be derived from classical laboratory experiment data, calculations in quantum mechanics, or both. Force fields utilize the same concept as force fields in classical physics, with the main difference that the force field parameters in chemistry describe the energy landscape on the atomistic level. From a force field, the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
Joel L. Sussman is an Israeli crystallographer best known for his studies on acetylcholinesterase, a key protein involved in transmission of nerve signals. He is the Morton and Gladys Pickman Professor of Structural Biology at the Weizmann Institute of Science in Rehovot and its director of the Israel Structural Proteomics Center.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.

UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.
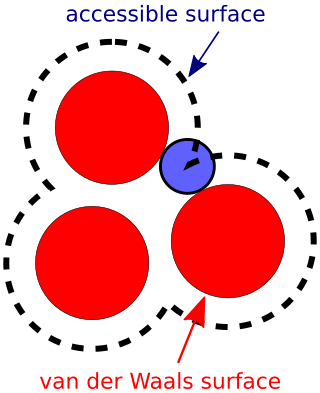
The accessible surface area (ASA) or solvent-accessible surface area (SASA) is the surface area of a biomolecule that is accessible to a solvent. Measurement of ASA is usually described in units of square angstroms. ASA was first described by Lee & Richards in 1971 and is sometimes called the Lee-Richards molecular surface. ASA is typically calculated using the 'rolling ball' algorithm developed by Shrake & Rupley in 1973. This algorithm uses a sphere of a particular radius to 'probe' the surface of the molecule.
BALL is a C++ class framework and set of algorithms and data structures for molecular modelling and computational structural bioinformatics, a Python interface to this library, and a graphical user interface to BALL, the molecule viewer BALLView.
Implicit solvation is a method to represent solvent as a continuous medium instead of individual “explicit” solvent molecules, most often used in molecular dynamics simulations and in other applications of molecular mechanics. The method is often applied to estimate free energy of solute-solvent interactions in structural and chemical processes, such as folding or conformational transitions of proteins, DNA, RNA, and polysaccharides, association of biological macromolecules with ligands, or transport of drugs across biological membranes.
In molecular modelling, docking is a method which predicts the preferred orientation of one molecule to another when bound together in a stable complex. In the case of protein docking, the search space consists of all possible orientations of the protein with respect to the ligand. Flexible docking in addition considers all possible conformations of the protein paired with all possible conformations of the ligand.
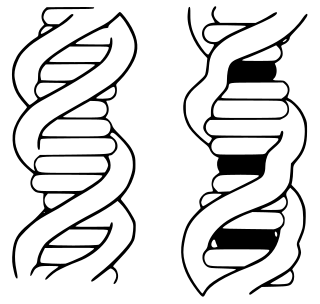
In biochemistry, intercalation is the insertion of molecules between the planar bases of deoxyribonucleic acid (DNA). This process is used as a method for analyzing DNA and it is also the basis of certain kinds of poisoning.
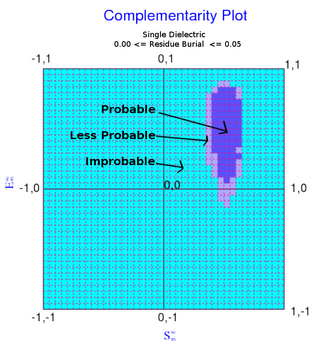
The complementarity plot (CP) is a graphical tool for structural validation of atomic models for both folded globular proteins and protein-protein interfaces. It is based on a probabilistic representation of preferred amino acid side-chain orientation, analogous to the preferred backbone orientation of Ramachandran plots). It can potentially serve to elucidate protein folding as well as binding. The upgraded versions of the software suite is available and maintained in github for both folded globular proteins as well as inter-protein complexes. The software is included in the bioinformatic tool suites OmicTools and Delphi tools.
References
- ↑ Katzir, Ephraim (2009). "Chapter 33". A Life's Tale (English language ed.). Carmel Publishing House. ISBN 978-965-540-026-7.
- ↑ Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA (1992). "Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques". Proc Natl Acad Sci USA. 89 (6): 2195–2199. Bibcode:1992PNAS...89.2195K. doi: 10.1073/pnas.89.6.2195 . PMC 48623 . PMID 1549581.
- ↑ "MolFit". Weizmann Institute of Science. Retrieved 22 February 2018.
- ↑ "FTDock (v2.0)". Structural Bioinformatics Group. Retrieved 22 February 2018.
| | This molecular modelling–related article is a stub. You can help Wikipedia by expanding it. |