| Kosaki Overgrowth Syndrome | |
|---|---|
 | |
| Kosaki overgrowth syndrome is inherited in an autosomal dominant manner. |

Kosaki overgrowth syndrome is a rare syndrome caused by mutations in the PDGFRB gene. [1]
| Kosaki Overgrowth Syndrome | |
|---|---|
| | |
| Kosaki overgrowth syndrome is inherited in an autosomal dominant manner. |
Kosaki overgrowth syndrome is a rare syndrome caused by mutations in the PDGFRB gene. [1]
The features of this syndrome affect the face, skin, brain and the body.[ citation needed ]
Face:
Skin:
Brain:
Body:
The height, lower-segment, hand, and foot length are all greater than usual.
No inheritance pattern has been described as these mutations appear to have arisen de novo. This syndrome is due to mutations in a single copy of the PDGFRB gene.[ citation needed ]
| | This section is empty. You can help by adding to it. (February 2018) |
This condition was first described in Japan in 2011 by Watanabe et al. [2] These authors thought the condition was the Shprintzen-Goldberg syndrome but the patient lacked a mutation in the SKI gene. A second case was described by Takenouchi et al in 2015. [3] These authors recognised that this condition was novel and on performing a whole genome sequencing found mutations in the PDGFRB gene. A further 24 cases were reported in 2017 by Gawliński et al. [4]

Waardenburg syndrome is a group of rare genetic conditions characterised by at least some degree of congenital hearing loss and pigmentation deficiencies, which can include bright blue eyes, a white forelock or patches of light skin. These basic features constitute type 2 of the condition; in type 1, there is also a wider gap between the inner corners of the eyes called telecanthus, or dystopia canthorum. In type 3, which is rare, the arms and hands are also malformed, with permanent finger contractures or fused fingers, while in type 4, the person also has Hirschsprung's disease. There also exist at least two types that can result in central nervous system (CNS) symptoms such as developmental delay and muscle tone abnormalities.

Alpha-thalassemia mental retardation syndrome (ATRX), also called alpha-thalassemia X-linked intellectual disability syndrome, nondeletion type or ATR-X syndrome, is an X-linked recessive condition associated with a mutation in the ATRX gene. Males with this condition tend to be moderately intellectually disabled and have physical characteristics including coarse facial features, microcephaly, hypertelorism, a depressed nasal bridge, a tented upper lip and an everted lower lip. Mild or moderate anemia, associated with alpha-thalassemia, is part of the condition. Females with this mutated gene have no specific signs or features, but if they do, they may demonstrate skewed X chromosome inactivation.
Acromicric dysplasia is an extremely rare inherited disorder characterized by abnormally short hands and feet, growth retardation and delayed bone maturation leading to short stature. Most cases have occurred randomly for no apparent reason (sporadically). However, autosomal dominant inheritance has not been ruled out.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.
Vici syndrome, also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum, is a rare autosomal recessive congenital disorder characterized by albinism, agenesis of the corpus callosum, cataracts, cardiomyopathy, severe psychomotor retardation, seizures, immunodeficiency and recurrent severe infections. To date, about 50 cases have been reported.

Fukutin is a eukaryotic protein necessary for the maintenance of muscle integrity, cortical histogenesis, and normal ocular development. Mutations in the fukutin gene have been shown to result in Fukuyama congenital muscular dystrophy (FCMD) characterised by brain malformation - one of the most common autosomal-recessive disorders in Japan. In humans this protein is encoded by the FCMD gene, located on chromosome 9q31. Human fukutin exhibits a length of 461 amino acids and a predicted molecular mass of 53.7 kDa.

Platelet-derived growth factor receptor beta is a protein that in humans is encoded by the PDGFRB gene. Mutations in PDGFRB are mainly associated with the clonal eosinophilia class of malignancies.

Phosphatidylinositol N-acetylglucosaminyltransferase subunit A is the catalytic subunit of the phosphatidylinositol N-acetylglucosaminyltransferase enzyme, which in humans is encoded by the PIGA gene.
Stiff skin syndrome is a cutaneous condition characterized by ‘rock hard’ induration, thickening of the skin and subcutaneous tissues, limited joint mobility, and mild hypertrichosis in infancy or early childhood. Immunologic abnormalities or vascular hyperactivity are not present in patients.

Parkes Weber syndrome (PWS) is a congenital disorder of the vascular system. It is an extremely rare condition, and its exact prevalence is unknown. It is named after British dermatologist Frederick Parkes Weber, who first described the syndrome in 1907.

Macrocephaly-capillary malformation (M-CM) is a multiple malformation syndrome causing abnormal body and head overgrowth and cutaneous, vascular, neurologic, and limb abnormalities. Though not every patient has all features, commonly found signs include macrocephaly, congenital macrosomia, extensive cutaneous capillary malformation, body asymmetry, polydactyly or syndactyly of the hands and feet, lax joints, doughy skin, variable developmental delay and other neurologic problems such as seizures and low muscle tone.
Shprintzen–Goldberg syndrome is a congenital multiple-anomaly syndrome that has craniosynostosis, multiple abdominal hernias, cognitive impairment, and other skeletal malformations as key features. Several reports have linked the syndrome to a mutation in the FBN1 gene, but these cases do not resemble those initially described in the medical literature in 1982 by Shprintzen and Goldberg, and Greally et al. in 1998 failed to find a causal link to FBN1. At this time, the cause of Shprintzen–Goldberg syndrome has been identified as a mutation in the gene SKI located on chromosome 1 at the p36 locus. The syndrome is rare with fewer than 50 cases described in the medical literature to date.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
Knobloch syndrome is a rare genetic disorder presenting severe eyesight problems and often a defect in the skull. It was named after the ophthalmologist William Hunter Knobloch, who first described the syndrome in 1971. A usual occurrence is a degeneration of the vitreous humour and the retina, two components of the eye. This breakdown often results in the separation of the retina from the eye, called retinal detachment, which can be recurrent. Extreme myopia (near-sightedness) is a common feature. The limited evidence available from electroretinography suggests that a cone-rod pattern of dysfunction is also a feature.
ZTTK syndrome is a rare disease caused in humans by a genetic mutation of the SON gene. Common symptoms include developmental delay and sometimes moderate to several intellectual disability.
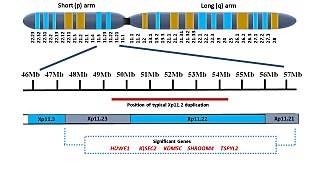
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).
Autosomal dominant intellectual disability-craniofacial anomalies-cardiac defects syndrome is a rare genetic disorder which is characterized by multi-systemic symptoms primarily affecting the intellect and post-natal development.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.

Salt and pepper developmental regression syndrome, also known as Amish infantile epileptic syndrome or GM3 deficiency syndrome, is a rare autosomal recessive progressive neurological disorder characterized by developmental delay, severe intellectual disability, seizures, and skin pigmentation irregularities. The clinical symptoms of this condition start manifesting soon after birth, during the newborn/neo-natal stage of life.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although it's phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.