Related Research Articles
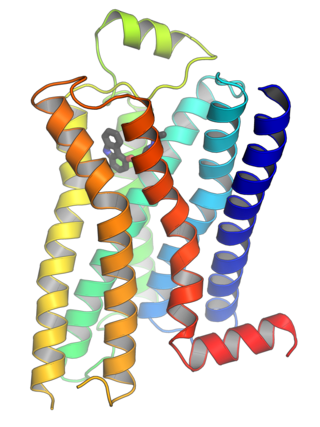
G protein-coupled receptors (GPCRs), also known as seven-(pass)-transmembrane domain receptors, 7TM receptors, heptahelical receptors, serpentine receptors, and G protein-linked receptors (GPLR), form a large group of evolutionarily related proteins that are cell surface receptors that detect molecules outside the cell and activate cellular responses. They are coupled with G proteins. They pass through the cell membrane seven times in form of six loops of amino acid residues, which is why they are sometimes referred to as seven-transmembrane receptors. Ligands can bind either to the extracellular N-terminus and loops or to the binding site within transmembrane helices. They are all activated by agonists, although a spontaneous auto-activation of an empty receptor has also been observed.

Integrins are transmembrane receptors that help cell-cell and cell-extracellular matrix (ECM) adhesion. Upon ligand binding, integrins activate signal transduction pathways that mediate cellular signals such as regulation of the cell cycle, organization of the intracellular cytoskeleton, and movement of new receptors to the cell membrane. The presence of integrins allows rapid and flexible responses to events at the cell surface.
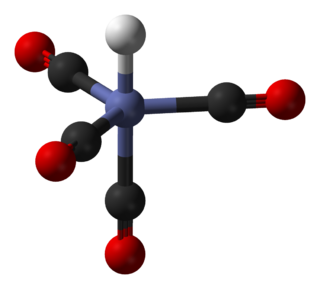
In coordination chemistry, a ligand is an ion or molecule with a functional group that binds to a central metal atom to form a coordination complex. The bonding with the metal generally involves formal donation of one or more of the ligand's electron pairs, often through Lewis bases. The nature of metal–ligand bonding can range from covalent to ionic. Furthermore, the metal–ligand bond order can range from one to three. Ligands are viewed as Lewis bases, although rare cases are known to involve Lewis acidic "ligands".

In biochemistry and pharmacology, receptors are chemical structures, composed of protein, that receive and transduce signals that may be integrated into biological systems. These signals are typically chemical messengers which bind to a receptor and produce physiological responses such as change in the electrical activity of a cell. For example, GABA, an inhibitory neurotransmitter inhibits electrical activity of neurons by binding to GABAA receptors. There are three main ways the action of the receptor can be classified: relay of signal, amplification, or integration. Relaying sends the signal onward, amplification increases the effect of a single ligand, and integration allows the signal to be incorporated into another biochemical pathway.
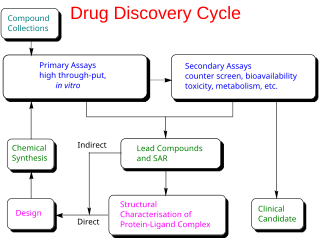
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.

Fas ligand is a type-II transmembrane protein expressed on cytotoxic T lymphocytes and natural killer (NK) cells. Its binding with Fas receptor (FasR) induces programmed cell death in the FasR-carrying target cell. Fas ligand/receptor interactions play an important role in the regulation of the immune system and the progression of cancer.

In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from Latin ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
In biology, cell signaling or cell communication is the ability of a cell to receive, process, and transmit signals with its environment and with itself. Cell signaling is a fundamental property of all cellular life in prokaryotes and eukaryotes. Signals that originate from outside a cell can be physical agents like mechanical pressure, voltage, temperature, light, or chemical signals. Cell signaling can occur over short or long distances, and as a result can be classified as autocrine, juxtacrine, intracrine, paracrine, or endocrine. Signaling molecules can be synthesized from various biosynthetic pathways and released through passive or active transports, or even from cell damage.
Protein–ligand docking is a molecular modelling technique. The goal of protein–ligand docking is to predict the position and orientation of a ligand when it is bound to a protein receptor or enzyme. Pharmaceutical research employs docking techniques for a variety of purposes, most notably in the virtual screening of large databases of available chemicals in order to select likely drug candidates. There has been rapid development in computational ability to determine protein structure with programs such as AlphaFold, and the demand for the corresponding protein-ligand docking predictions is driving implementation of software that can find accurate models. Once the protein folding can be predicted accurately along with how the ligands of various structures will bind to the protein, the ability for drug development to progress at a much faster rate becomes possible.
The retinoid X receptor (RXR) is a type of nuclear receptor that is activated by 9-cis retinoic acid, which is discussed controversially to be of endogenous relevance, and 9-cis-13,14-dihydroretinoic acid, which is likely to be the major endogenous mammalian RXR-selective agonist.
The formyl peptide receptors (FPR) belong to a class of G protein-coupled receptors involved in chemotaxis. In humans, there are three formyl peptide receptor isoforms, each encoded by a separate gene that are named FPR1, FPR2, and FPR3. These receptors were originally identified by their ability to bind N-formyl peptides such as N-formylmethionine produced by the degradation of either bacterial or host cells. Hence formyl peptide receptors are involved in mediating immune cell response to infection. These receptors may also act to suppress the immune system under certain conditions. The close phylogenetic relation of signaling in chemotaxis and olfaction was recently proved by detection formyl peptide receptor like proteins as a distinct family of vomeronasal organ chemosensors in mice.
In molecular modelling, docking is a method which predicts the preferred orientation of one molecule to another when bound together in a stable complex. In the case of protein docking, the search space consists of all possible orientations of the protein with respect to the ligand. Flexible docking in addition considers all possible conformations of the protein paired with all possible conformations of the ligand.

Arthur Mallay Lesk, is a protein science researcher, who is a professor of biochemistry and molecular biology at the Pennsylvania State University in University Park.

Ribbon diagrams, also known as Richardson diagrams, are 3D schematic representations of protein structure and are one of the most common methods of protein depiction used today. The ribbon depicts the general course and organisation of the protein backbone in 3D and serves as a visual framework for hanging details of the entire atomic structure, such as the balls for the oxygen atoms attached to myoglobin's active site in the adjacent figure. Ribbon diagrams are generated by interpolating a smooth curve through the polypeptide backbone. α-helices are shown as coiled ribbons or thick tubes, β-strands as arrows, and non-repetitive coils or loops as lines or thin tubes. The direction of the polypeptide chain is shown locally by the arrows, and may be indicated overall by a colour ramp along the length of the ribbon.

Ephrin A1 is a protein that in humans is encoded by the EFNA1 gene.
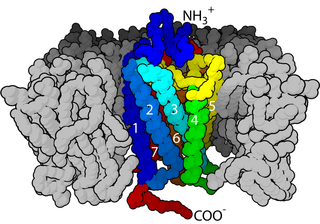
Cell surface receptors are receptors that are embedded in the plasma membrane of cells. They act in cell signaling by receiving extracellular molecules. They are specialized integral membrane proteins that allow communication between the cell and the extracellular space. The extracellular molecules may be hormones, neurotransmitters, cytokines, growth factors, cell adhesion molecules, or nutrients; they react with the receptor to induce changes in the metabolism and activity of a cell. In the process of signal transduction, ligand binding affects a cascading chemical change through the cell membrane.

NKG2D is an activating receptor (transmembrane protein) belonging to the NKG2 family of C-type lectin-like receptors. NKG2D is encoded by KLRK1 (killer cell lectin like receptor K1) gene which is located in the NK-gene complex (NKC) situated on chromosome 6 in mice and chromosome 12 in humans. In mice, it is expressed by NK cells, NK1.1+ T cells, γδ T cells, activated CD8+ αβ T cells and activated macrophages. In humans, it is expressed by NK cells, γδ T cells and CD8+ αβ T cells. NKG2D recognizes induced-self proteins from MIC and RAET1/ULBP families which appear on the surface of stressed, malignant transformed, and infected cells.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
LeDock is a simple proprietary molecular docking software that can be used for docking of ligands with protein targets. LeDock supports running on 64-bit Linux, macOS, and 32-bit and 64-bit Windows.
References
- ↑ Wallace AC, Laskowski RA, Thornton JM (February 1995). "LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions". Protein Eng. 8 (2): 127–34. doi:10.1093/protein/8.2.127. PMID 7630882.
| | This molecular physics–related article is a stub. You can help Wikipedia by expanding it. |