In chemistry, the oxidation state, or oxidation number, is the hypothetical charge of an atom if all of its bonds to other atoms were fully ionic. It describes the degree of oxidation of an atom in a chemical compound. Conceptually, the oxidation state may be positive, negative or zero. While fully ionic bonds are not found in nature, many bonds exhibit strong ionicity, making oxidation state a useful predictor of charge.

In computer science, the clique problem is the computational problem of finding cliques in a graph. It has several different formulations depending on which cliques, and what information about the cliques, should be found. Common formulations of the clique problem include finding a maximum clique, finding a maximum weight clique in a weighted graph, listing all maximal cliques, and solving the decision problem of testing whether a graph contains a clique larger than a given size.
In theoretical computer science, the subgraph isomorphism problem is a computational task in which two graphs G and H are given as input, and one must determine whether G contains a subgraph that is isomorphic to H. Subgraph isomorphism is a generalization of both the maximum clique problem and the problem of testing whether a graph contains a Hamiltonian cycle, and is therefore NP-complete. However certain other cases of subgraph isomorphism may be solved in polynomial time.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.

In the mathematical area of graph theory, a clique is a subset of vertices of an undirected graph such that every two distinct vertices in the clique are adjacent. That is, a clique of a graph is an induced subgraph of that is complete. Cliques are one of the basic concepts of graph theory and are used in many other mathematical problems and constructions on graphs. Cliques have also been studied in computer science: the task of finding whether there is a clique of a given size in a graph is NP-complete, but despite this hardness result, many algorithms for finding cliques have been studied.
A chemical database is a database specifically designed to store chemical information. This information is about chemical and crystal structures, spectra, reactions and syntheses, and thermophysical data.
In mathematics, computer science and especially graph theory, a distance matrix is a square matrix containing the distances, taken pairwise, between the elements of a set. Depending upon the application involved, the distance being used to define this matrix may or may not be a metric. If there are N elements, this matrix will have size N×N. In graph-theoretic applications, the elements are more often referred to as points, nodes or vertices.

OpenEye Scientific Software is an American software company founded by Anthony Nicholls in 1997. It develops large-scale molecular modelling applications and toolkits. Following OpenEye's acquisition by Cadence Design Systems for $500 million in September 2022, the company was rebranded to OpenEye Cadence Molecular Sciences and operates as a business unit under Cadence.

The uranyl ion is an oxycation of uranium in the oxidation state +6, with the chemical formula UO2+
2. It has a linear structure with short U–O bonds, indicative of the presence of multiple bonds between uranium and oxygen. Four or more ligands may be bound to the uranyl ion in an equatorial plane around the uranium atom. The uranyl ion forms many complexes, particularly with ligands that have oxygen donor atoms. Complexes of the uranyl ion are important in the extraction of uranium from its ores and in nuclear fuel reprocessing.
This page describes mining for molecules. Since molecules may be represented by molecular graphs this is strongly related to graph mining and structured data mining. The main problem is how to represent molecules while discriminating the data instances. One way to do this is chemical similarity metrics, which has a long tradition in the field of cheminformatics.
Protein–ligand docking is a molecular modelling technique. The goal of protein–ligand docking is to predict the position and orientation of a ligand when it is bound to a protein receptor or enzyme. Pharmaceutical research employs docking techniques for a variety of purposes, most notably in the virtual screening of large databases of available chemicals in order to select likely drug candidates. There has been rapid development in computational ability to determine protein structure with programs such as AlphaFold, and the demand for the corresponding protein-ligand docking predictions is driving implementation of software that can find accurate models. Once the protein folding can be predicted accurately along with how the ligands of various structures will bind to the protein, the ability for drug development to progress at a much faster rate becomes possible.
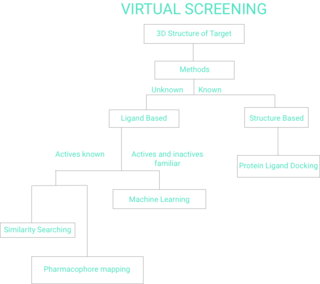
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
This list of structural comparison and alignment software is a compilation of software tools and web portals used in pairwise or multiple structural comparison and structural alignment.

Chemical similarity refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in inorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds.
Computer Atlas of Surface Topography of Proteins (CASTp) aims to provide comprehensive and detailed quantitative characterization of topographic features of protein, is now updated to version 3.0. Since its release in 2006, the CASTp server has ≈45000 visits and fulfills ≈33000 calculation requests annually. CASTp has been proven as a confident tool for a wide range of researches, including investigations of signaling receptors, discoveries of cancer therapeutics, understanding of mechanism of drug actions, studies of immune disorder diseases, analysis of protein–nanoparticle interactions, inference of protein functions and development of high-throughput computational tools. This server is maintained by Jie Liang's lab in University of Illinois at Chicago.
Early twenty-first century pesticide research has focused on developing molecules that combine low use rates and that are more selective, safer, resistance-breaking and cost-effective. Obstacles include increasing pesticide resistance and an increasingly stringent regulatory environment.

The MaxCliqueDyn algorithm is an algorithm for finding a maximum clique in an undirected graph.
ProBiS is a computer software which allows prediction of binding sites and their corresponding ligands for a given protein structure. Initially ProBiS was developed as a ProBiS algorithm by Janez Konc and Dušanka Janežič in 2010 and is now available as ProBiS server, ProBiS CHARMMing server, ProBiS algorithm and ProBiS plugin. The name ProBiS originates from the purpose of the software itself, that is to predict for a given Protein structure Binding Sites and their corresponding ligands.

