Related Research Articles
A chemical database is a database specifically designed to store chemical information. This information is about chemical and crystal structures, spectra, reactions and syntheses, and thermophysical data.
Cheminformatics refers to the use of physical chemistry theory with computer and information science techniques—so called "in silico" techniques—in application to a range of descriptive and prescriptive problems in the field of chemistry, including in its applications to biology and related molecular fields. Such in silico techniques are used, for example, by pharmaceutical companies and in academic settings to aid and inform the process of drug discovery, for instance in the design of well-defined combinatorial libraries of synthetic compounds, or to assist in structure-based drug design. The methods can also be used in chemical and allied industries, and such fields as environmental science and pharmacology, where chemical processes are involved or studied.
Chemical Markup Language is an approach to managing molecular information using tools such as XML and Java. It was the first domain specific implementation based strictly on XML, first based on a DTD and later on an XML Schema, the most robust and widely used system for precise information management in many areas. It has been developed over more than a decade by Murray-Rust, Rzepa and others and has been tested in many areas and on a variety of machines.
The International Chemical Identifier is a textual identifier for chemical substances, designed to provide a standard way to encode molecular information and to facilitate the search for such information in databases and on the web. Initially developed by the International Union of Pure and Applied Chemistry (IUPAC) and National Institute of Standards and Technology (NIST) from 2000 to 2005, the format and algorithms are non-proprietary. Since May 2009, it has been developed by the InChI Trust, a nonprofit charity from the United Kingdom which works to implement and promote the use of InChI.
Open Babel is computer software, a chemical expert system mainly used to interconvert chemical file formats.
JOELib is computer software, a chemical expert system used mainly to interconvert chemical file formats. Because of its strong relationship to informatics, this program belongs more to the category cheminformatics than to molecular modelling. It is available for Windows, Unix and other operating systems supporting the programming language Java. It is free and open-source software distributed under the GNU General Public License (GPL) 2.0.

The Chemistry Development Kit (CDK) is computer software, a library in the programming language Java, for chemoinformatics and bioinformatics. It is available for Windows, Linux, Unix, and macOS. It is free and open-source software distributed under the GNU Lesser General Public License (LGPL) 2.0.

Henry Stephen Rzepa is a chemist and Emeritus Professor of Computational Chemistry at Imperial College London.

Peter Murray-Rust is a chemist currently working at the University of Cambridge. As well as his work in chemistry, Murray-Rust is also known for his support of open access and open data.

The Bioclipse project is a Java-based, open-source, visual platform for chemo- and bioinformatics based on the Eclipse Rich Client Platform (RCP).

The JME Molecule Editor is a molecule editor Java applet with which users make and edit drawings of molecules and reactions, and can display molecules within an HTML page. The editor can generate Daylight simplified molecular-input line-entry system (SMILES) or MDL Molfiles of the created structures.
Molecular descriptors play a fundamental role in chemistry, pharmaceutical sciences, environmental protection policy, and health researches, as well as in quality control, being the way molecules, thought of as real bodies, are transformed into numbers, allowing some mathematical treatment of the chemical information contained in the molecule. This was defined by Todeschini and Consonni as:
SMILES arbitrary target specification (SMARTS) is a language for specifying substructural patterns in molecules. The SMARTS line notation is expressive and allows extremely precise and transparent substructural specification and atom typing.

Chemical similarity refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in inorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds.

JChemPaint is computer software, a molecule editor and file viewer for chemical structures using 2D computer graphics. It is free and open-source software, released under a GNU Lesser General Public License (LGPL). It is written in Java and so can run on the operating systems Windows, macOS, Linux, and Unix. There is a standalone application (editor), and two varieties of applet that can be integrated into web pages.
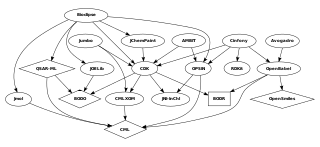
Blue Obelisk is an informal group of chemists who promote open data, open source, and open standards; it was initiated by Peter Murray-Rust and others in 2005. Multiple open source cheminformatics projects associate themselves with the Blue Obelisk, among which, in alphabetical order, Avogadro, Bioclipse, cclib, Chemistry Development Kit, GaussSum, JChemPaint, JOELib, Kalzium, Openbabel, OpenSMILES, and UsefulChem.

The Journal of Cheminformatics is a peer-reviewed open access scientific journal that covers cheminformatics and molecular modelling. It was established in 2009 with David Wild and Christoph Steinbeck as founding editors-in-chief, and was originally published by Chemistry Central. At the end of 2015, the Chemistry Central brand was retired and its titles, including Journal of Cheminformatics, were merged with the SpringerOpen portfolio of open access journals.

Christoph Steinbeck is a German chemist and has a professorship for analytical chemistry, cheminformatics and chemometrics at the Friedrich-Schiller-Universität Jena in Thuringia.
Matched molecular pair analysis (MMPA) is a method in cheminformatics that compares the properties of two molecules that differ only by a single chemical transformation, such as the substitution of a hydrogen atom by a chlorine one. Such pairs of compounds are known as matched molecular pairs (MMP). Because the structural difference between the two molecules is small, any experimentally observed change in a physical or biological property between the matched molecular pair can more easily be interpreted. The term was first coined by Kenny and Sadowski in the book Chemoinformatics in Drug Discovery.
A chemical graph generator is a software package to generate computer representations of chemical structures adhering to certain boundary conditions. The development of such software packages is a research topic of cheminformatics. Chemical graph generators are used in areas such as virtual library generation in drug design, in molecular design with specified properties, called inverse QSAR/QSPR, as well as in organic synthesis design, retrosynthesis or in systems for computer-assisted structure elucidation (CASE). CASE systems again have regained interest for the structure elucidation of unknowns in computational metabolomics, a current area of computational biology.
References
- ↑ Jean-Loup Faulon; Andreas Bender (April 2010). Handbook of Chemoinformatics Algorithms. Chapman & Hall. ISBN 978-1420082920.
- ↑ Johann Gasteiger (November 2003). Chemoinformatics . Wiley-VCH. ISBN 3527306811.
- ↑ Steinbeck C, C.; Han Y; Kuhn S; Horlacher O; Luttmann E; Willighagen E (2003). "The Chemistry Development Kit". J. Chem. Inf. Comput. Sci. 43 (2): 493–500. doi:10.1021/ci025584y. PMC 4901983 . PMID 12653513.
- ↑ Steinbeck C., Christoph; Hoppe C.; Kuhn S.; Floris M.; Guha R.; Willighagen E.L. (2006). "Recent Developments of the Chemistry Development Kit (CDK) - An Open-Source Java Library for Chemo- and Bioinformatics". Curr. Pharm. Des. 12 (17): 2111–2120. doi:10.2174/138161206777585274. hdl: 2066/35445 . PMID 16796559.
- ↑ Willighagen, Egon L.; Mayfield, John W.; Alvarsson, Jonathan; Berg, Arvid; Carlsson, Lars; Jeliazkova, Nina; Kuhn, Stefan; Pluskal, Tomáš; Rojas-Chertó, Miquel; Spjuth, Ola; Torrance, Gilleain; Evelo, Chris T.; Guha, Rajarshi; Steinbeck, Christoph (December 2017). "The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching". Journal of Cheminformatics. 9 (1): 33. doi: 10.1186/s13321-017-0220-4 . PMC 5461230 . PMID 29086040. S2CID 2019985.
- ↑ reads and writes all chemical file formats.
- ↑ O’Boyle N; Banck M; James C; Morley C; Vandermeersch T; Hutchison G (2011). "Open babel: an open chemical". Journal of Cheminformatics. 3 (33): 33. doi: 10.1186/1758-2946-3-33 . PMC 3198950 . PMID 21982300.