Related Research Articles
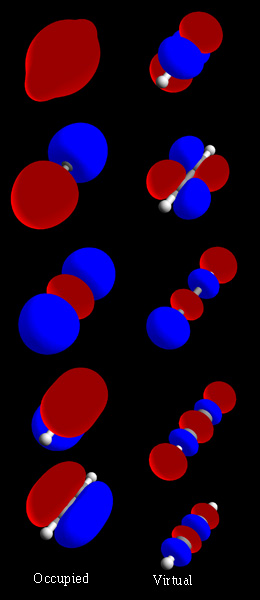
In chemistry, a molecular orbital is a mathematical function describing the location and wave-like behavior of an electron in a molecule. This function can be used to calculate chemical and physical properties such as the probability of finding an electron in any specific region. The terms atomic orbital and molecular orbital were introduced by Robert S. Mulliken in 1932 to mean one-electron orbital wave functions. At an elementary level, they are used to describe the region of space in which a function has a significant amplitude.
A linear combination of atomic orbitals or LCAO is a quantum superposition of atomic orbitals and a technique for calculating molecular orbitals in quantum chemistry. In quantum mechanics, electron configurations of atoms are described as wavefunctions. In a mathematical sense, these wave functions are the basis set of functions, the basis functions, which describe the electrons of a given atom. In chemical reactions, orbital wavefunctions are modified, i.e. the electron cloud shape is changed, according to the type of atoms participating in the chemical bond.
In computational physics and chemistry, the Hartree–Fock (HF) method is a method of approximation for the determination of the wave function and the energy of a quantum many-body system in a stationary state.
The Roothaan equations are a representation of the Hartree–Fock equation in a non orthonormal basis set which can be of Gaussian-type or Slater-type. It applies to closed-shell molecules or atoms where all molecular orbitals or atomic orbitals, respectively, are doubly occupied. This is generally called Restricted Hartree–Fock theory.

Vladimir Aleksandrovich Fock was a Soviet physicist, who did foundational work on quantum mechanics and quantum electrodynamics.
Psi is an ab initio computational chemistry package originally written by the research group of Henry F. Schaefer, III. Utilizing Psi, one can perform a calculation on a molecular system with various kinds of methods such as Hartree-Fock, Post-Hartree–Fock electron correlation methods, and density functional theory. The program can compute energies, optimize molecular geometries, and compute vibrational frequencies. The major part of the program is written in C++, while Python API is also available, which allows users to perform complex computations or automate tasks easily.
Møller–Plesset perturbation theory (MP) is one of several quantum chemistry post–Hartree–Fock ab initio methods in the field of computational chemistry. It improves on the Hartree–Fock method by adding electron correlation effects by means of Rayleigh–Schrödinger perturbation theory (RS-PT), usually to second (MP2), third (MP3) or fourth (MP4) order. Its main idea was published as early as 1934 by Christian Møller and Milton S. Plesset.
Configuration interaction (CI) is a post-Hartree–Fock linear variational method for solving the nonrelativistic Schrödinger equation within the Born–Oppenheimer approximation for a quantum chemical multi-electron system. Mathematically, configuration simply describes the linear combination of Slater determinants used for the wave function. In terms of a specification of orbital occupation (for instance, (1s)2(2s)2(2p)1...), interaction means the mixing (interaction) of different electronic configurations (states). Due to the long CPU time and large memory required for CI calculations, the method is limited to relatively small systems.
Electronic correlation is the interaction between electrons in the electronic structure of a quantum system. The correlation energy is a measure of how much the movement of one electron is influenced by the presence of all other electrons.
Multi-configurational self-consistent field (MCSCF) is a method in quantum chemistry used to generate qualitatively correct reference states of molecules in cases where Hartree–Fock and density functional theory are not adequate. It uses a linear combination of configuration state functions (CSF), or configuration determinants, to approximate the exact electronic wavefunction of an atom or molecule. In an MCSCF calculation, the set of coefficients of both the CSFs or determinants and the basis functions in the molecular orbitals are varied to obtain the total electronic wavefunction with the lowest possible energy. This method can be considered a combination between configuration interaction and Hartree–Fock.
In computational chemistry, post–Hartree–Fock (post-HF) methods are the set of methods developed to improve on the Hartree–Fock (HF), or self-consistent field (SCF) method. They add electron correlation which is a more accurate way of including the repulsions between electrons than in the Hartree–Fock method where repulsions are only averaged.
Koopmans' theorem states that in closed-shell Hartree–Fock theory (HF), the first ionization energy of a molecular system is equal to the negative of the orbital energy of the highest occupied molecular orbital (HOMO). This theorem is named after Tjalling Koopmans, who published this result in 1934.
In quantum mechanics, the exchange operator, also known as permutation operator, is a quantum mechanical operator that acts on states in Fock space. The exchange operator acts by switching the labels on any two identical particles described by the joint position quantum state . Since the particles are identical, the notion of exchange symmetry requires that the exchange operator be unitary.
Unrestricted Hartree–Fock (UHF) theory is the most common molecular orbital method for open shell molecules where the number of electrons of each spin are not equal. While restricted Hartree–Fock theory uses a single molecular orbital twice, one multiplied by the α spin function and the other multiplied by the β spin function in the Slater determinant, unrestricted Hartree–Fock theory uses different molecular orbitals for the α and β electrons. This has been called a different orbitals for different spins (DODS) method. The result is a pair of coupled Roothaan equations, known as the Pople–Nesbet–Berthier equations.
Restricted open-shell Hartree–Fock (ROHF) is a variant of Hartree–Fock method for open shell molecules. It uses doubly occupied molecular orbitals as far as possible and then singly occupied orbitals for the unpaired electrons. This is the simple picture for open shell molecules but it is difficult to implement. The foundations of the ROHF method were first formulated by Clemens C. J. Roothaan in a celebrated paper and then extended by various authors, see e.g. for in-depth discussions.
Computational chemical methods in solid-state physics follow the same approach as they do for molecules, but with two differences. First, the translational symmetry of the solid has to be utilised, and second, it is possible to use completely delocalised basis functions such as plane waves as an alternative to the molecular atom-centered basis functions. The electronic structure of a crystal is in general described by a band structure, which defines the energies of electron orbitals for each point in the Brillouin zone. Ab initio and semi-empirical calculations yield orbital energies, therefore they can be applied to band structure calculations. Since it is time-consuming to calculate the energy for a molecule, it is even more time-consuming to calculate them for the entire list of points in the Brillouin zone.
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initio was first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene. The background is described by Parr. Ab initio means "from first principles" or "from the beginning", implying that the only inputs into an ab initio calculation are physical constants. Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order to yield useful information such as electron densities, energies and other properties of the system. The ability to run these calculations has enabled theoretical chemists to solve a range of problems and their importance is highlighted by the awarding of the Nobel prize to John Pople and Walter Kohn.
Understanding the structure of the atomic nucleus is one of the central challenges in nuclear physics.
In computer software, FreeON is an experimental, open source (GPL) suite of programs for linear scaling quantum chemistry, formerly known as MondoSCF. It is highly modular, and has been written from scratch for N-scaling SCF theory in Fortran95 and C. Platform independent IO is supported with HDF5. FreeON should compile with most modern Linux distributions. FreeON performs Hartree–Fock, pure density functional, and hybrid HF/DFT calculations in a Cartesian-Gaussian LCAO basis. All algorithms are O(N) or O(N lg N) for non-metallic systems. Periodic boundary conditions in 1, 2 and 3 dimensions have been implemented through the Lorentz field, and an internal coordinate geometry optimizer allows full (atom+cell) relaxation using analytic derivatives. Effective core potentials for energies and forces have been implemented, but Effective Core Potential (ECP) lattice forces do not work yet. Advanced features include O(N) static and dynamic response, as well as time reversible Born Oppenheimer Molecular Dynamics (MD).
Hartree is the unit of energy in the Hartree atomic units system.