
Brachydactyly is a medical term denoting the presence of abnormally short digits at birth. The shortness is relative to the length of other long bones and other parts of the body. Brachydactyly is an inherited, dominant trait. It most often occurs as an isolated dysmelia, but can also occur with other anomalies as part of many congenital syndromes. Brachydactyly may also be a signal that one is at risk for congenital heart disease due to the association between congenital heart disease and Carpenter syndrome and the link between Carpenter syndrome and brachydactyly.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Turricephaly is a type of cephalic disorder where the head appears tall with a small length and width. It is due to premature closure of the coronal suture plus any other suture, like the lambdoid, or it may be used to describe the premature fusion of all sutures. It should be differentiated from Crouzon syndrome. Oxycephaly is a form of turricephaly where the head is cone-shaped, and is the most severe of the craniosynostoses.

Nephritis is inflammation of the kidneys and may involve the glomeruli, tubules, or interstitial tissue surrounding the glomeruli and tubules. It is one of several different types of nephropathy.

Schmitt Gillenwater Kelly syndrome is a rare autosomal dominant congenital disorder consisting of radial hypoplasia, triphalangeal thumbs, hypospadias, and maxillary diastema.

Sabinas brittle hair syndrome, also called Sabinas syndrome or brittle hair-mental deficit syndrome, is an autosomal recessive congenital disorder affecting the integumentary system.

Autoimmune polyendocrine syndromes (APSs), also called polyglandular autoimmune syndromes (PGASs) or polyendocrine autoimmune syndromes (PASs), are a heterogeneous group of rare diseases characterized by autoimmune activity against more than one endocrine organ, although non-endocrine organs can be affected. There are three types of APS, and there are a number of other diseases which involve endocrine autoimmunity.
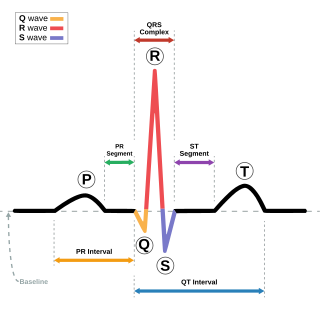
Romano–Ward syndrome is the most common form of congenital Long QT syndrome (LQTS), a genetic heart condition that affects the electrical properties of heart muscle cells. Those affected are at risk of abnormal heart rhythms which can lead to fainting, seizures, or sudden death. Romano–Ward syndrome can be distinguished clinically from other forms of inherited LQTS as it affects only the electrical properties of the heart, while other forms of LQTS can also affect other parts of the body.
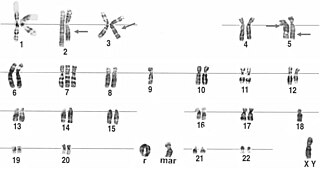
A ring chromosome is an aberrant chromosome whose ends have fused together to form a ring. Ring chromosomes were first discovered by Lilian Vaughan Morgan in 1926. A ring chromosome is denoted by the symbol r in human genetics and R in Drosophila genetics. Ring chromosomes may form in cells following genetic damage by mutagens like radiation, but they may also arise spontaneously during development.
Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.
TNF receptor associated periodic syndrome (TRAPS) is a periodic fever syndrome associated with mutations in a receptor for the molecule tumor necrosis factor (TNF) that is inheritable in an autosomal dominant manner. Individuals with TRAPS have episodic symptoms such as recurrent high fevers, rash, abdominal pain, joint/muscle aches and puffy eyes.

RAPADILINO syndrome is an autosomal recessive disorder characterized by:
Naegeli–Franceschetti–Jadassohn syndrome (NFJS), also known as chromatophore nevus of Naegeli and Naegeli syndrome, is a rare autosomal dominant form of ectodermal dysplasia, characterized by reticular skin pigmentation, diminished function of the sweat glands, the absence of teeth and hyperkeratosis of the palms and soles. One of the most striking features is the absence of fingerprint lines on the fingers.
Congenital contractural arachnodactyly (CCA), also known as Beals–Hecht syndrome, is a rare autosomal dominant congenital connective tissue disorder. As with Marfan syndrome, people with CCA typically have an arm span that is greater than their height and very long fingers and toes. However, Beals and Hecht discovered in 1972 that, unlike Marfan's, CCA is caused by mutations to the fibrillin-2 (FBN2) gene rather than the fibrillin-1 (FBN1) gene.

In medical contexts, a facies is a distinctive facial expression or appearance associated with a specific medical condition. The term comes from Latin for "face". As a fifth declension noun, facies can be both singular and plural.
Björnstad syndrome is an autosomal recessive congenital condition involving pili torti, sensorineural deafness, and hair abnormalities. It was first characterized in 1965, in Oslo, by prof. Roar Theodor Bjørnstad after he observed an association between pili torti and hearing loss. The condition is extremely rare, with less than 50 cases documented in medical literature worldwide.
Ulnar–mammary syndrome or Schinzel syndrome is a cutaneous condition characterized by nipple and breast hypoplasia, i.e. underdevelopment. Features of UMS can be mild to severe and can vary significantly from person to person, even within the same family. The main features of UMS include upper limb defects, underdevelopment of the apocrine and mammary glands, and various genital abnormalities. Other signs and symptoms may include hormonal deficiencies, delayed puberty, dental problems and obesity. People with UMS may have distinct facial features, including a wide face tapering to a prominent chin, and a broad nose.

Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive or , visual impairment from early adulthood onwards and dementia from the 4th decade onwards. The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.
Nablus mask-like facial syndrome is a rare genetic condition. It is a microdeletion syndrome triggered by a deletion at chromosome 8 q22.1 that causes a mask-like facial appearance in those affected. This syndrome typically presents itself in infants, specifically newborns.
