
The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.

The SIB Swiss Institute of Bioinformatics is an academic not-for-profit foundation which federates bioinformatics activities throughout Switzerland.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.

In molecular biology, an intrinsically disordered protein (IDP) is a protein that lacks a fixed or ordered three-dimensional structure, typically in the absence of its macromolecular interaction partners, such as other proteins or RNA. IDPs range from fully unstructured to partially structured and include random coil, molten globule-like aggregates, or flexible linkers in large multi-domain proteins. They are sometimes considered as a separate class of proteins along with globular, fibrous and membrane proteins.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.
In molecular biology and genetics, transcription coregulators are proteins that interact with transcription factors to either activate or repress the transcription of specific genes. Transcription coregulators that activate gene transcription are referred to as coactivators while those that repress are known as corepressors. The mechanism of action of transcription coregulators is to modify chromatin structure and thereby make the associated DNA more or less accessible to transcription. In humans several dozen to several hundred coregulators are known, depending on the level of confidence with which the characterisation of a protein as a coregulator can be made. One class of transcription coregulators modifies chromatin structure through covalent modification of histones. A second ATP dependent class modifies the conformation of chromatin.
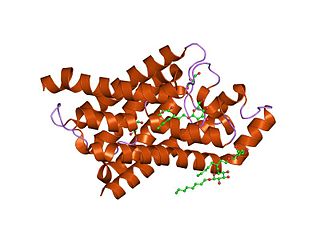
Major intrinsic proteins comprise a large superfamily of transmembrane protein channels that are grouped together on the basis of homology. The MIP superfamily includes three subfamilies: aquaporins, aquaglyceroporins and S-aquaporins.
- The aquaporins (AQPs) are water selective.
- The aquaglyceroporins are permeable to water, but also to other small uncharged molecules such as glycerol.
- The third subfamily, with little conserved amino acid sequences around the NPA boxes, include 'superaquaporins' (S-aquaporins).

Lens fiber major intrinsic protein also known as aquaporin-0 is a protein that in humans is encoded by the MIP gene.
The Proteolysis MAP (PMAP) was an integrated web resource focused on proteases. Its domain now links to a scam/spam browser extender.

In molecular biology short linear motifs (SLiMs), linear motifs or minimotifs are short stretches of protein sequence that mediate protein–protein interaction.
DisProt is a manually curated biological database of intrinsically disordered proteins (IDPs) and regions (IDRs). DisProt annotations cover state information on the protein but also, when available, its state transitions, interactions and functional aspects of disorder detected by specific experimental methods. DisProt is hosted and maintained in the BioComputing UP laboratory.

Prokaryotic ubiquitin-like protein (Pup) is a functional analog of ubiquitin found in the prokaryote Mycobacterium tuberculosis. Like ubiquitin, Pup serves to direct proteins to the proteasome for degradation in the Pup-proteasome system (PPS). However, the enzymology of ubiquitylation and pupylation is different, owing to their distinct evolutionary origins. In contrast to the three-step reaction of ubiquitylation, pupylation requires only two steps, and thus only two enzymes are involved in pupylation. The enzymes involved in pupylation are descended from glutamine synthetase.

In molecular biology, protein fold classes are broad categories of protein tertiary structure topology. They describe groups of proteins that share similar amino acid and secondary structure proportions. Each class contains multiple, independent protein superfamilies.
Molecular recognition features (MoRFs) are small intrinsically disordered regions in proteins that undergo a disorder-to-order transition upon binding to their partners. MoRFs are implicated in protein-protein interactions, which serve as the initial step in molecular recognition. MoRFs are disordered prior to binding to their partners, whereas they form a common 3D structure after interacting with their partners. As MoRF regions tend to resemble disordered proteins with some characteristics of ordered proteins, they can be classified as existing in an extended semi-disordered state.

In computational chemistry, conformational ensembles, also known as structural ensembles, are experimentally constrained computational models describing the structure of intrinsically unstructured proteins. Such proteins are flexible in nature, lacking a stable tertiary structure, and therefore cannot be described with a single structural representation. The techniques of ensemble calculation are relatively new on the field of structural biology, and are still facing certain limitations that need to be addressed before it will become comparable to classical structural description methods such as biological macromolecular crystallography.
In molecular biology, MobiDB is a curated biological database designed to offer a centralized resource for annotations of intrinsic protein disorder. Protein disorder is a structural feature characterizing a large number of proteins with prominent members known as intrinsically unstructured proteins. The database features three levels of annotation: manually curated, indirect and predicted. By combining different data sources of protein disorder into a consensus annotation, MobiDB aims at giving the best possible picture of the "disorder landscape" of a given protein of interest.
Ramasubbu Sankararamakrishnan is an Indian computational biologist, bioinformatician and a professor at the Department of Biological Sciences and Bioengineering of the Indian Institute of Technology, Kanpur. He is known for his computational studies on membrane protein function. The Department of Biotechnology of the Government of India awarded him the National Bioscience Award for Career Development, one of the highest Indian science awards, for his contributions to biosciences in 2008.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.