X-ray crystallography is the experimental science of determining the atomic and molecular structure of a crystal, in which the crystalline structure causes a beam of incident X-rays to diffract in specific directions. By measuring the angles and intensities of the X-ray diffraction, a crystallographer can produce a three-dimensional picture of the density of electrons within the crystal and the positions of the atoms, as well as their chemical bonds, crystallographic disorder, and other information.
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules such as proteins and nucleic acids, which is overseen by the Worldwide Protein Data Bank (wwPDB). These structural data are obtained and deposited by biologists and biochemists worldwide through the use of experimental methodologies such as X-ray crystallography, NMR spectroscopy, and, increasingly, cryo-electron microscopy. All submitted data are reviewed by expert biocurators and, once approved, are made freely available on the Internet under the CC0 Public Domain Dedication. Global access to the data is provided by the websites of the wwPDB member organisations.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
BioJava is an open-source software project dedicated to provide Java tools to process biological data. BioJava is a set of library functions written in the programming language Java for manipulating sequences, protein structures, file parsers, Common Object Request Broker Architecture (CORBA) interoperability, Distributed Annotation System (DAS), access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a Protein Data Bank (PDB) file, interacting with Jmol and many more. This application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis.
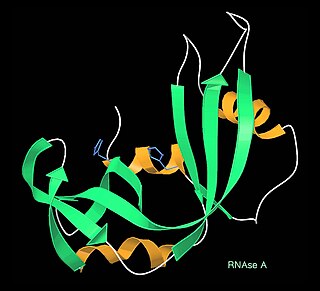
A kinemage is an interactive graphic scientific illustration. It often is used to visualize molecules, especially proteins although it can also represent other types of 3-dimensional data. The kinemage system is designed to optimize ease of use, interactive performance, and the perception and communication of detailed 3D information. The kinemage information is stored in a text file, human- and machine-readable, that describes the hierarchy of display objects and their properties, and includes optional explanatory text. The kinemage format is a defined chemical MIME type of 'chemical/x-kinemage' with the file extension '.kin'.
A chemical file format is a type of data file which is used specifically for depicting molecular data. One of the most widely used is the chemical table file format, which is similar to Structure Data Format (SDF) files. They are text files that represent multiple chemical structure records and associated data fields. The XYZ file format is a simple format that usually gives the number of atoms in the first line, a comment on the second, followed by a number of lines with atomic symbols and cartesian coordinates. The Protein Data Bank Format is commonly used for proteins but is also used for other types of molecules. There are many other types which are detailed below. Various software systems are available to convert from one format to another.
The Protein Data Bank (PDB) file format is a textual file format describing the three-dimensional structures of molecules held in the Protein Data Bank, now succeeded by the mmCIF format. The PDB format accordingly provides for description and annotation of protein and nucleic acid structures including atomic coordinates, secondary structure assignments, as well as atomic connectivity. In addition experimental metadata are stored. The PDB format is the legacy file format for the Protein Data Bank which has kept data on biological macromolecules in the newer PDBx/mmCIF file format since 2014.

Crystallographic Information File (CIF) is a standard text file format for representing crystallographic information, promulgated by the International Union of Crystallography (IUCr). CIF was developed by the IUCr Working Party on Crystallographic Information in an effort sponsored by the IUCr Commission on Crystallographic Data and the IUCr Commission on Journals. The file format was initially published by Hall, Allen, and Brown and has since been revised, most recently versions 1.1 and 2.0. Full specifications for the format are available at the IUCr website. Many computer programs for molecular viewing are compatible with this format, including Jmol.
The EM Data Bank or Electron Microscopy Data Bank (EMDB) collects 3D EM maps and associated experimental data determined using electron microscopy of biological specimens. It was established in 2002 at the MSD/PDBe group of the European Bioinformatics Institute (EBI), where the European site of the EMDataBank.org consortium is located. As of 2015, the resource contained over 2,600 entries with a mean resolution of 15Å.

The Cambridge Structural Database (CSD) is both a repository and a validated and curated resource for the three-dimensional structural data of molecules generally containing at least carbon and hydrogen, comprising a wide range of organic, metal-organic and organometallic molecules. The specific entries are complementary to the other crystallographic databases such as the Protein Data Bank (PDB), Inorganic Crystal Structure Database and International Centre for Diffraction Data. The data, typically obtained by X-ray crystallography and less frequently by electron diffraction or neutron diffraction, and submitted by crystallographers and chemists from around the world, are freely accessible on the Internet via the CSD's parent organization's website. The CSD is overseen by the not-for-profit incorporated company called the Cambridge Crystallographic Data Centre, CCDC.

Helen Miriam Berman is a Board of Governors Professor of Chemistry and Chemical Biology at Rutgers University and a former director of the RCSB Protein Data Bank. A structural biologist, her work includes structural analysis of protein-nucleic acid complexes, and the role of water in molecular interactions. She is also the founder and director of the Nucleic Acid Database, and led the Protein Structure Initiative Structural Genomics Knowledgebase.
In biology, a protein structure database is a database that is modeled around the various experimentally determined protein structures. The aim of most protein structure databases is to organize and annotate the protein structures, providing the biological community access to the experimental data in a useful way. Data included in protein structure databases often includes three-dimensional coordinates as well as experimental information, such as unit cell dimensions and angles for x-ray crystallography determined structures. Though most instances, in this case either proteins or a specific structure determinations of a protein, also contain sequence information and some databases even provide means for performing sequence based queries, the primary attribute of a structure database is structural information, whereas sequence databases focus on sequence information, and contain no structural information for the majority of entries. Protein structure databases are critical for many efforts in computational biology such as structure based drug design, both in developing the computational methods used and in providing a large experimental dataset used by some methods to provide insights about the function of a protein.
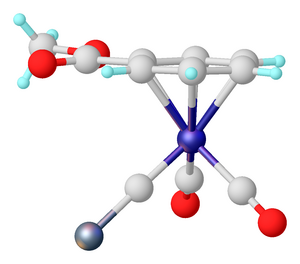
A crystallographic database is a database specifically designed to store information about the structure of molecules and crystals. Crystals are solids having, in all three dimensions of space, a regularly repeating arrangement of atoms, ions, or molecules. They are characterized by symmetry, morphology, and directionally dependent physical properties. A crystal structure describes the arrangement of atoms, ions, or molecules in a crystal.

The program Coot is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics. It is primarily focused on building and validation of atomic models into three-dimensional electron density maps obtained by X-ray crystallography methods, although it has also been applied to data from electron microscopy.
Cryo bio-crystallography is the application of crystallography to biological macromolecules at cryogenic temperatures.

The Protein Common Interface Database (ProtCID) is a database of similar protein-protein interfaces in crystal structures of homologous proteins.
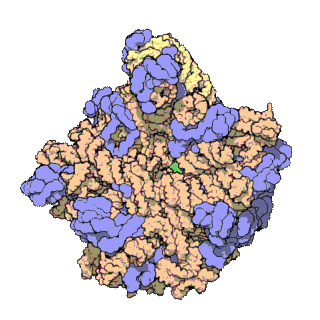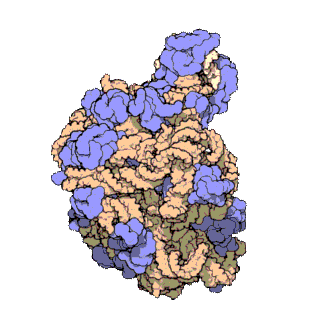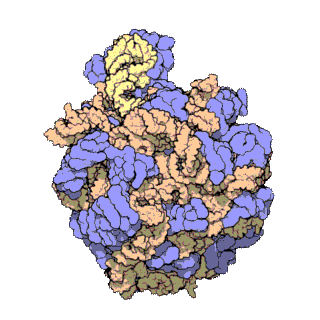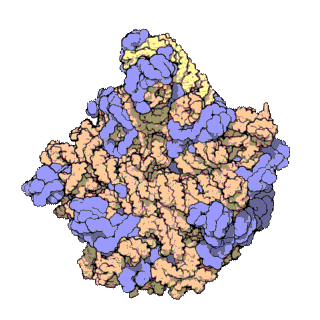
The term macromolecular assembly (MA) refers to massive chemical structures such as viruses and non-biologic nanoparticles, cellular organelles and membranes and ribosomes, etc. that are complex mixtures of polypeptide, polynucleotide, polysaccharide or other polymeric macromolecules. They are generally of more than one of these types, and the mixtures are defined spatially, and with regard to their underlying chemical composition and structure. Macromolecules are found in living and nonliving things, and are composed of many hundreds or thousands of atoms held together by covalent bonds; they are often characterized by repeating units. Assemblies of these can likewise be biologic or non-biologic, though the MA term is more commonly applied in biology, and the term supramolecular assembly is more often applied in non-biologic contexts. MAs of macromolecules are held in their defined forms by non-covalent intermolecular interactions, and can be in either non-repeating structures, or in repeating linear, circular, spiral, or other patterns. The process by which MAs are formed has been termed molecular self-assembly, a term especially applied in non-biologic contexts. A wide variety of physical/biophysical, chemical/biochemical, and computational methods exist for the study of MA; given the scale of MAs, efforts to elaborate their composition and structure and discern mechanisms underlying their functions are at the forefront of modern structure science.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
The Biological Magnetic Resonance Data Bank is an open access repository of nuclear magnetic resonance (NMR) spectroscopic data from peptides, proteins, nucleic acids and other biologically relevant molecules. The database is operated by the University of Wisconsin–Madison and is supported by the National Library of Medicine. The BMRB is part of the Research Collaboratory for Structural Bioinformatics and, since 2006, it is a partner in the Worldwide Protein Data Bank (wwPDB). The repository accepts NMR spectral data from laboratories around the world and, once the data is validated, it is available online at the BMRB website. The database has also an ftp site, where data can be downloaded in the bulk. The BMRB has two mirror sites, one at the Protein Database Japan (PDBj) at Osaka University and one at the Magnetic Resonance Research Center (CERM) at the University of Florence in Italy. The site at Japan accepts and processes data depositions.

Mercury is a freeware developed by the Cambridge Crystallographic Data Centre, originally designed as a crystal structure visualization tool. Mercury helps three dimensional visualization of crystal structure and assists in drawing and analysis of crystal packing and intermolecular interactions. Current version Mercury can read "cif", ".mol", ".mol2", ".pdb", ".res", ".sd" and ".xyz" types of files. Mercury has its own file format with filename extension ".mryx".