Related Research Articles

The regulation of therapeutic goods, defined as drugs and therapeutic devices, varies by jurisdiction. In some countries, such as the United States, they are regulated at the national level by a single agency. In other jurisdictions they are regulated at the state level, or at both state and national levels by various bodies, as in Australia.

The European Medicines Agency (EMA) is an agency of the European Union (EU) in charge of the evaluation and supervision of pharmaceutical products. Prior to 2004, it was known as the European Agency for the Evaluation of Medicinal Products or European Medicines Evaluation Agency (EMEA).
The Medicines and Healthcare products Regulatory Agency (MHRA) is an executive agency of the Department of Health and Social Care in the United Kingdom which is responsible for ensuring that medicines and medical devices work and are acceptably safe.
The Veterinary Medicines Directorate (VMD) is an Executive Agency of the Department for Environment, Food and Rural Affairs (Defra) seeking to protect public health, animal health, the environment and promoting animal welfare by assuring the safety, quality and efficacy of veterinary medicines in the United Kingdom.

In the European Economic Area, a supplementary protection certificate (SPC) is a sui generis intellectual property (IP) right that extends the duration of certain rights associated with a patent. It enters into force after expiry of a patent upon which it is based. This type of right is available for various regulated, biologically active agents, namely human or veterinary medicaments and plant protection products. Supplementary protection certificates were introduced to encourage innovation by compensating for the long time needed to obtain regulatory approval of these products.
Test data exclusivity refers to protection of clinical trial data required to be submitted to a regulatory agency to prove safety and efficacy of a new drug, and prevention of generic drug manufacturers from relying on this data in their own applications. It provides a form of market exclusivity outside that provided by patent rights.

The European Pharmacopoeia is a major regional pharmacopoeia which provides common quality standards throughout the pharmaceutical industry in Europe to control the quality of medicines, and the substances used to manufacture them. It is a published collection of monographs which describe both the individual and general quality standards for ingredients, dosage forms, and methods of analysis for medicines. These standards apply to medicines for both human and veterinary use.
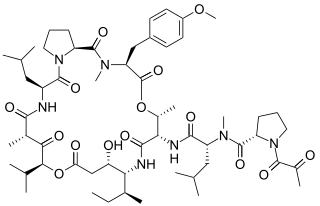
Plitidepsin is a chemical compound extracted from the ascidian Aplidium albicans. It is currently undergoing clinical trial testing. It is a member of the class of compounds known as didemnins.

EudraLex is the collection of rules and regulations governing medicinal products in the European Union.
The Clinical Trials Directive is a European Union directive that aimed at facilitating the internal market in medicinal products within the European Union, while at the same time maintaining an appropriate level of protection for public health. It seeks to simplify and harmonise the administrative provisions governing clinical trials in the European Community, by establishing a clear, transparent procedure.
Ustekinumab, sold under the brand name Stelara among others, is a monoclonal antibody medication developed by Janssen Pharmaceuticals, for the treatment of Crohn's disease, ulcerative colitis, plaque psoriasis and psoriatic arthritis, targeting both IL-12 and IL-23.
The European Directive on Traditional Herbal Medicinal Products (THMPD), formally the Directive 2004/24/EC amending, as regards traditional herbal medicinal products, Directive 2001/83/EC on the Community code relating to medicinal products for human use, was established by the European Parliament and Council on 31 March 2004 to provide a simplified regulatory approval process for traditional herbal medicines in the European Union (EU). Previously, there was no formal EU wide authorisation procedure, so each EU member state regulated these types of products at the national level.
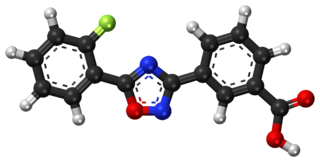
Ataluren, sold under the brand name Translarna, is a medication for the treatment of Duchenne muscular dystrophy. It was designed by PTC Therapeutics.

Garenoxacin (INN) is a quinolone antibiotic for the treatment of Gram-positive and Gram-negative bacterial infections.

Pradofloxacin, sold under the brand name Veraflox among others, is a third-generation enhanced spectrum veterinary antibiotic of the fluoroquinolone class. It was developed by Elanco Animal Health GmbH and received approval from the European Commission in April 2011, for prescription-only use in veterinary medicine for the treatment of bacterial infections in dogs and cats.
In the European Union, the Qualified Person Responsible For Pharmacovigilance (QPPV) is an individual, usually an employee of a pharmaceutical company, who is personally responsible for the safety of the human pharmaceutical products marketed by that company in the EU. This function was established in 2004 by article 23 of regulation (EC) No 726/2004. The article establishes that the holder of a marketing authorization for a drug for human use must have a QPPV. When a company submits an application for permission to bring a medicinal product onto the market, the company submits a description of its system for monitoring the safety of the product in actual use and proof that the services of a QPPV are in place.
Olaratumab, sold under the brand name Lartruvo, is a monoclonal antibody medication developed by Eli Lilly and Company for the treatment of solid tumors. It is directed against the platelet-derived growth factor receptor alpha.
Marketing authorisation is the process of reviewing and assessing the evidence to support a medicinal product, such as a drug, in relation to its marketing, finalised by granting of a licence to be sold.
Paediatric-use marketing authorisations (PUMA) are granted by the European Medicines Agency (EMA) for medical products that are intended exclusively for paediatric use, that is, for use in patients younger than 18 years. Like ordinary EMA marketing authorisations, a PUMA approval is valid in all countries of the European Economic Area. The PUMA process was established to make it more profitable for pharmaceutical companies to market drugs for children. For this purpose, new data used for PUMA approved drugs are protected for 10 years, and the applications are partially exempt from fees.

Valneva COVID-19 vaccine is a COVID-19 vaccine developed by French biotechnology company Valneva SE in collaboration with the American biopharmaceutical company Dynavax Technologies.