| | |
|---|---|
| Content | |
| Description | DNA methylation data. |
| Contact | |
| Primary citation | PMID 11125109 |
| Access | |
| Website | http://www.methdb.de |
MethDB is a database for DNA methylation data. [1]
| | |
|---|---|
| Content | |
| Description | DNA methylation data. |
| Contact | |
| Primary citation | PMID 11125109 |
| Access | |
| Website | http://www.methdb.de |
MethDB is a database for DNA methylation data. [1]
In the chemical sciences, methylation denotes the addition of a methyl group on a substrate, or the substitution of an atom by a methyl group. Methylation is a form of alkylation, with a methyl group replacing a hydrogen atom. These terms are commonly used in chemistry, biochemistry, soil science, and the biological sciences.

The CpG sites or CG sites are regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases along its 5' → 3' direction. CpG sites occur with high frequency in genomic regions called CpG islands. Cytosines in CpG dinucleotides can be methylated to form 5-methylcytosines. Enzymes that add a methyl group are called DNA methyltransferases. In mammals, 70% to 80% of CpG cytosines are methylated. Methylating the cytosine within a gene can change its expression, a mechanism that is part of a larger field of science studying gene regulation that is called epigenetics.
In genetics, an expressed sequence tag (EST) is a short sub-sequence of a cDNA sequence. ESTs may be used to identify gene transcripts, and are instrumental in gene discovery and in gene-sequence determination. The identification of ESTs has proceeded rapidly, with approximately 74.2 million ESTs now available in public databases.

Regulation of gene expression, or gene regulation, includes a wide range of mechanisms that are used by cells to increase or decrease the production of specific gene products. Sophisticated programs of gene expression are widely observed in biology, for example to trigger developmental pathways, respond to environmental stimuli, or adapt to new food sources. Virtually any step of gene expression can be modulated, from transcriptional initiation, to RNA processing, and to the post-translational modification of a protein. Often, one gene regulator controls another, and so on, in a gene regulatory network.
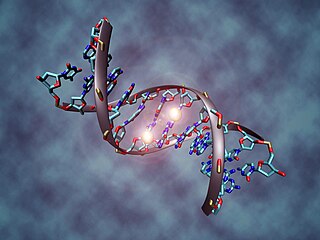
DNA methylation is a biological process by which methyl groups are added to the DNA molecule. Methylation can change the activity of a DNA segment without changing the sequence. When located in a gene promoter, DNA methylation typically acts to repress gene transcription. In mammals, DNA methylation is essential for normal development and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, repression of transposable elements, aging, and carcinogenesis.

Methyltransferases are a large group of enzymes that all methylate their substrates but can be split into several subclasses based on their structural features. The most common class of methyltransferases is class I, all of which contain a Rossmann fold for binding S-Adenosyl methionine (SAM). Class II methyltransferases contain a SET domain, which are exemplified by SET domain histone methyltransferases, and class III methyltransferases, which are membrane associated. Methyltransferases can also be grouped as different types utilizing different substrates in methyl transfer reactions. These types include protein methyltransferases, DNA/RNA methyltransferases, natural product methyltransferases, and non-SAM dependent methyltransferases. SAM is the classical methyl donor for methyltrasferases, however, examples of other methyl donors are seen in nature. The general mechanism for methyl transfer is a SN2-like nucleophilic attack where the methionine sulfur serves as the nucleophile that transfers the methyl group to the enzyme substrate. SAM is converted to S-Adenosyl homocysteine (SAH) during this process. The breaking of the SAM-methyl bond and the formation of the substrate-methyl bond happen nearly simultaneously. These enzymatic reactions are found in many pathways and are implicated in genetic diseases, cancer, and metabolic diseases. Another type of methyl transfer is the radical S-Adenosyl methionine (SAM) which is the methylation of unactivated carbon atoms in primary metabolites, proteins, lipids, and RNA.

Bisulfitesequencing (also known as bisulphite sequencing) is the use of bisulfite treatment of DNA before routine sequencing to determine the pattern of methylation. DNA methylation was the first discovered epigenetic mark, and remains the most studied. In animals it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG, and is implicated in repression of transcriptional activity.
SNORD29 is a non-coding RNA (ncRNA) molecule which functions in the modification of other small nuclear RNAs (snRNAs). This type of modifying RNA is usually located in the nucleolus of the eukaryotic cell which is a major site of snRNA biogenesis. It is known as a small nucleolar RNA (snoRNA) and also often referred to as a guide RNA.

snoRNA U41 belongs to the C/D box class of snoRNAs. It is predicted to guide 2'O-ribose methylation of the large 28S rRNA on residue U4276.
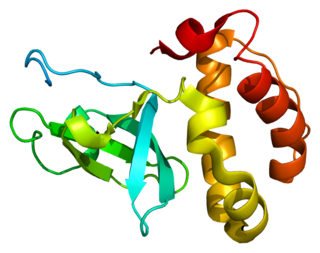
DNA (cytosine-5-)-methyltransferase 3 beta, is an enzyme that in humans in encoded by the DNMT3B gene. Mutation in this gene are associated with immunodeficiency, centromere instability and facial anomalies syndrome.
PubMeth is a database that contains information about DNA hypermethylation in cancer. It can be queried either by searching a list of genes, or cancer (sub)types.

2′-O-methylation is a common nucleoside modification of RNA, where a methyl group is added to the 2′ hydroxyl of the ribose moiety of a nucleoside, producing a methoxy group. 2′-O-methylated nucleosides are mostly found in ribosomal RNA and small nuclear RNA and occur in the functionally essential regions of the ribosome and spliceosome. Currently, about 1210 2′-O-methylations (2′-O-Me) have been identified in mammals and yeast and deposited in RMBase database.
Computational epigenetics uses bioinformatic methods to complement experimental research in epigenetics. Due to the recent explosion of epigenome datasets, computational methods play an increasing role in all areas of epigenetic research.

The Single Nucleotide Polymorphism Database (dbSNP) is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI). Although the name of the database implies a collection of one class of polymorphisms only, it in fact contains a range of molecular variation: (1) SNPs, (2) short deletion and insertion polymorphisms (indels/DIPs), (3) microsatellite markers or short tandem repeats (STRs), (4) multinucleotide polymorphisms (MNPs), (5) heterozygous sequences, and (6) named variants. The dbSNP accepts apparently neutral polymorphisms, polymorphisms corresponding to known phenotypes, and regions of no variation. It was created in September 1998 to supplement GenBank, NCBI’s collection of publicly available nucleic acid and protein sequences.
The Illumina Methylation Assay using the Infinium I platform uses 'BeadChip' technology to generate a comprehensive genome-wide profiling of human DNA methylation. Similar to bisulfite sequencing and pyrosequencing, this method quantifies methylation levels at various loci within the genome. This assay is used for methylation probes on the Illumina Infinium HumanMethylation27 BeadChip. Probes on the 27k array target regions of the human genome to measure methylation levels at 27,578 CpG dinucleotides in 14,495 genes. The Infinium HumanMethylation450 BeadChip array targets >450,000 methylation sites.
NGSmethDB is a database of methylation data derived from next-generation sequencing data.

Biomarkers of aging are biomarkers that could predict functional capacity at some later age better than chronological age. Stated another way, biomarkers of aging would give the true "biological age", which may be different from the chronological age.
MethBase is a database of DNA methylation data derived from next-generation sequencing data. MethBase provides a visualization of publicly available bisulfite sequencing and reduced representation bisulfite sequencing experiments through the UCSC Genome Browser. MethBase contents include single-CpG site resolution methylation levels for each CpG site in the genome of interest, annotation of regions of hypomethylation often associated with gene promoters, and annotation of allele-specific methylation associated with genomic imprinting.

An epigenome-wide association study (EWAS) is an examination of a genome-wide set of quantifiable epigenetic marks, such as DNA methylation, in different individuals to derive associations between epigenetic variation and a particular identifiable phenotype/trait. When patterns change such as DNA methylation at specific loci, discriminating the phenotypically affected cases from control individuals, this is considered an indication that epigenetic perturbation has taken place that is associated, causally or consequentially, with the phenotype.
| | This Biological database-related article is a stub. You can help Wikipedia by expanding it. |