Last updated The potential curve of the Mie potential in reduced units, for different values of the repulsive exponent (), all depicted curves use the attractive exponent . The black curve corresponds to the Lennard-Jones potential.
The Mie potential is an interaction potential describing the interactions between particles on the atomic level. It is mostly used for describing intermolecular interactions, but at times also for modeling intramolecular interaction, i.e. bonds.
The Mie potential is named after the German physicist Gustav Mie;[1] yet the history of intermolecular potentials is more complicated.[2][3][4] The Mie potential is the generalized case of the Lennard-Jones (LJ) potential, which is perhaps the most widely used pair potential.[5][6]
The Mie potential is a function of , the distance between two particles, and is written as[7]
with
such that the potential minimum is .
The Lennard-Jones potential corresponds to the special case where and in Eq. (1). In Eq. (1), is the dispersion energy, and indicates the distance at which , which is sometimes called the "collision radius." The parameter is generally indicative of the size of the particles involved in the collision. The parameters and characterize the shape of the potential: describes the character of the repulsion and describes the character of the attraction.
The attractive exponent is physically justified by the London dispersion force,[4] whereas no justification for a certain value for the repulsive exponent is known. The repulsive steepness parameter has a significant influence on the modeling of thermodynamic derivative properties, e.g. the compressibility and the speed of sound. Therefore, the Mie potential is a more flexible intermolecular potential than the simpler Lennard-Jones potential.
The Mie potential is used today in many force fields in molecular modeling. Typically, the attractive exponent is chosen to be , whereas the repulsive exponent is used as an adjustable parameter during the model fitting.
Thermophysical properties of the Mie substance
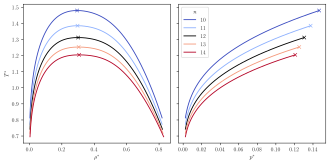
The reducedphase diagram of a fluid consisting of particles interacting through a Mie potential with different values for the repulsive exponent (), all with the attractive exponent . The cross indicates the critical point.
As for the Lennard-Jonesium, where a theoretical substance exists that is defined by particles interacting by the Lennard-Jones potential, a substance class of Mie substances exists that are defined as single site spherical particles interacting by a given Mie potential. Since an infinite number of Mie potentials exist (using different n, m parameters), equally many Mie substances exist, as opposed to Lennard-Jonesium, which is uniquely defined. For practical applications in molecular modelling, the Mie substances are mostly relevant for modelling small molecules, e.g. noble gases, and for coarse grain modelling, where larger molecules, or even a collection of molecules, are simplified in their structure and described by a single Mie particle. However, more complex molecules, such as long-chained alkanes, have successfully been modelled as homogeneous chains of Mie particles.[8] As such, the Mie potential is useful for modelling far more complex systems than those whose behaviour is accurately captured by "free" Mie particles.
Thermophysical properties of both the Mie fluid, and chain molecules built from Mie particles have been the subject of numerous papers in recent years. Investigated properties include virial coefficients[9] and interfacial,[10]vapor-liquid equilibrium,[11][12][13][14] and transport properties.[15] Based on such studies the relation between the shape of the interaction potential (described by n and m) and the thermophysical properties has been elucidated.
Also, many theoretical (analytical) models have been developed for describing thermophysical properties of Mie substances and chain molecules formed from Mie particles, such as several thermodynamic equations of state[8][16][17] and models for transport properties.[18]
It has been observed that many combinations of different () can yield similar phase behaviour,[19] and that this degeneracy is captured by the parameter
,
where fluids with different exponents, but the same -parameter will exhibit the same phase behavior.[19]
Mie potential used in molecular modeling
Due to its flexibility, the Mie potential is a popular choice for modelling real fluids in force fields. It is used as an interaction potential many molecular models today. Several (reliable) united atom transferable force fields are based on the Mie potential, such as that developed by Potoff and co-workers.[20][21][22] The Mie potential has also been used for coarse-grain modeling.[23] Electronic tools are available for building Mie force field models for both united atom force fields and transferable force fields.[24][23] The Mie potential has also been used for modeling small spherical molecules (i.e. directly the Mie substance - see above). The Table below gives some examples. There, the molecular models have only the parameters of the Mie potential itself.
↑ Lenhard, Johannes; Stephan, Simon; Hasse, Hans (February 2024). "A child of prediction. On the History, Ontology, and Computation of the Lennard-Jonesium". Studies in History and Philosophy of Science. 103: 105–113. doi:10.1016/j.shpsa.2023.11.007. ISSN0039-3681. PMID38128443. S2CID266440296.
↑ Pohl, Sven; Fingerhut, Robin; Thol, Monika; Vrabec, Jadran; Span, Roland (2023-02-27). "Equation of state for the Mie (λr,6) fluid with a repulsive exponent from 11 to 13". The Journal of Chemical Physics. 158 (8). doi:10.1063/5.0133412. ISSN0021-9606. PMID36859099. S2CID257249977.
1 2 Aasen, Ailo; Hammer, Morten; Ervik, Åsmund; Müller, Erich A.; Wilhelmsen, Øivind (2019-08-13). "Equation of state and force fields for Feynman–Hibbs-corrected Mie fluids. I. Application to pure helium, neon, hydrogen, and deuterium". The Journal of Chemical Physics. 151 (6). Bibcode:2019JChPh.151f4508A. doi:10.1063/1.5111364. hdl:10044/1/72226. ISSN0021-9606. S2CID202083098.
↑ Mick, Jason R.; Soroush Barhaghi, Mohammad; Jackman, Brock; Rushaidat, Kamel; Schwiebert, Loren; Potoff, Jeffrey J. (2015-09-16). "Optimized Mie potentials for phase equilibria: Application to noble gases and their mixtures with n-alkanes". The Journal of Chemical Physics. 143 (11). Bibcode:2015JChPh.143k4504M. doi:10.1063/1.4930138. ISSN0021-9606. PMID26395716. S2CID43211598.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.