Related Research Articles
GROMACS is a molecular dynamics package mainly designed for simulations of proteins, lipids, and nucleic acids. It was originally developed in the Biophysical Chemistry department of University of Groningen, and is now maintained by contributors in universities and research centers worldwide. GROMACS is one of the fastest and most popular software packages available, and can run on central processing units (CPUs) and graphics processing units (GPUs). It is free, open-source software released under the GNU General Public License (GPL), and starting with version 4.6, the GNU Lesser General Public License (LGPL).
Nanoscale Molecular Dynamics is computer software for molecular dynamics simulation, written using the Charm++ parallel programming model. It is noted for its parallel efficiency and is often used to simulate large systems. It has been developed by the collaboration of the Theoretical and Computational Biophysics Group (TCB) and the Parallel Programming Laboratory (PPL) at the University of Illinois at Urbana–Champaign.

Scientific visualization is an interdisciplinary branch of science concerned with the visualization of scientific phenomena. It is also considered a subset of computer graphics, a branch of computer science. The purpose of scientific visualization is to graphically illustrate scientific data to enable scientists to understand, illustrate, and glean insight from their data. Research into how people read and misread various types of visualizations is helping to determine what types and features of visualizations are most understandable and effective in conveying information.
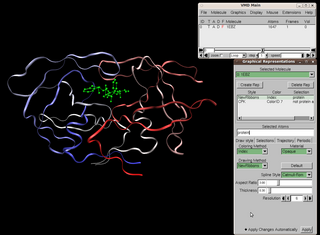
Visual Molecular Dynamics (VMD) is a molecular modelling and visualization computer program. VMD is developed mainly as a tool to view and analyze the results of molecular dynamics simulations. It also includes tools for working with volumetric data, sequence data, and arbitrary graphics objects. Molecular scenes can be exported to external rendering tools such as POV-Ray, RenderMan, Tachyon, Virtual Reality Modeling Language (VRML), and many others. Users can run their own Tcl and Python scripts within VMD as it includes embedded Tcl and Python interpreters. VMD runs on Unix, Apple Mac macOS, and Microsoft Windows. VMD is available to non-commercial users under a distribution-specific license which permits both use of the program and modification of its source code, at no charge.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.

The FOX toolkit is an open-source, cross-platform widget toolkit, i.e. a library of basic elements for building a graphical user interface (GUI). FOX stands for Free Objects for X.
ROOT is an object-oriented computer program and library developed by CERN. It was originally designed for particle physics data analysis and contains several features specific to the field, but it is also used in other applications such as astronomy and data mining. The latest minor release is 6.28, as of 2023-02-03.
ScientificPython is an open source library of scientific tools for the Python programming language. Its development started in 1995.
A chemical file format is a type of data file which is used specifically for depicting molecular data. One of the most widely used is the chemical table file format, which is similar to Structure Data Format (SDF) files. They are text files that represent multiple chemical structure records and associated data fields. The XYZ file format is a simple format that usually gives the number of atoms in the first line, a comment on the second, followed by a number of lines with atomic symbols and cartesian coordinates. The Protein Data Bank Format is commonly used for proteins but is also used for other types of molecules. There are many other types which are detailed below. Various software systems are available to convert from one format to another.
PyMOL is an open source but proprietary molecular visualization system created by Warren Lyford DeLano. It was commercialized initially by DeLano Scientific LLC, which was a private software company dedicated to creating useful tools that become universally accessible to scientific and educational communities. It is currently commercialized by Schrödinger, Inc. As the original software license was a permissive licence, they were able to remove it; new versions are no longer released under the Python license, but under a custom license, and some of the source code is no longer released. PyMOL can produce high-quality 3D images of small molecules and biological macromolecules, such as proteins. According to the original author, by 2009, almost a quarter of all published images of 3D protein structures in the scientific literature were made using PyMOL.

The Blender Game Engine was a free and open-source 3D production suite used for making real-time interactive content. It was previously embedded within Blender, but support for it was dropped in 2019, with the release of Blender 2.8. The game engine was written from scratch in C++ as a mostly independent component, and includes support for features such as Python scripting and OpenAL 3D sound.
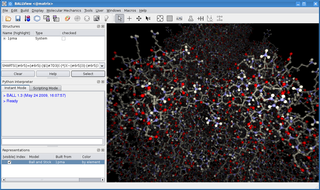
BALL is a C++ class framework and set of algorithms and data structures for molecular modelling and computational structural bioinformatics, a Python interface to this library, and a graphical user interface to BALL, the molecule viewer BALLView.
Tinker, previously stylized as TINKER, is a suite of computer software applications for molecular dynamics simulation. The codes provide a complete and general set of tools for molecular mechanics and molecular dynamics, with some special features for biomolecules. The core of the software is a modular set of callable routines which allow manipulating coordinates and evaluating potential energy and derivatives via straightforward means.

Computational Engineering is an emerging discipline that deals with the development and application of computational models for engineering, known as Computational Engineering Models or CEM. At this time, various different approaches are summarized under the term Computational Engineering, including using computational geometry and virtual design for engineering tasks, often coupled with a simulation-driven approach In Computational Engineering, algorithms solve mathematical and logical models that describe engineering challenges, sometimes coupled with some aspect of AI, specifically Reinforcement Learning.
Sirius is a molecular modelling and analysis system developed at San Diego Supercomputer Center. Sirius is designed to support advanced user requirements that go beyond simple display of small molecules and proteins. Sirius supports high quality interactive 3D graphics, structure building, displaying protein or DNA primary sequences, access to remote data sources, and visualizing molecular dynamics trajectories. It can be used for scientific visualization and analysis, and chemistry and biology instruction.
This is a list of computer programs that are predominantly used for molecular mechanics calculations.
Web-based simulation (WBS) is the invocation of computer simulation services over the World Wide Web, specifically through a web browser. Increasingly, the web is being looked upon as an environment for providing modeling and simulation applications, and as such, is an emerging area of investigation within the simulation community.
References
- 1 2 Hinsen K (2000). "The Molecular Modeling Toolkit: A New Approach to Molecular Simulations". J. Comput. Chem. 21 (2): 79–85. doi:10.1002/(SICI)1096-987X(20000130)21:2<79::AID-JCC1>3.0.CO;2-B.