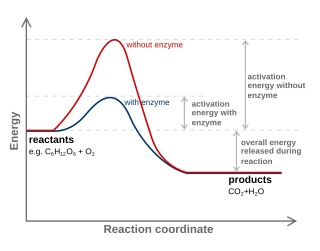
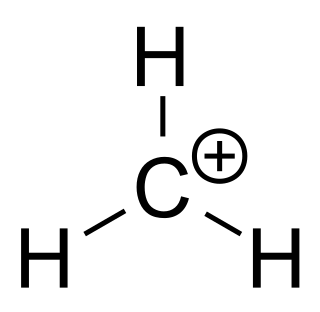
Catalysis is the process of increasing the rate of a chemical reaction by adding a substance known as a catalyst, which is not consumed in the catalyzed reaction and can continue to act repeatedly. Because of this, only very small amounts of catalyst are required to alter the reaction rate in principle.

The transition state of a chemical reaction is a particular configuration along the reaction coordinate. It is defined as the state corresponding to the highest potential energy along this reaction coordinate. At this point, assuming a perfectly irreversible reaction, colliding reactant molecules always go on to form products. It is often marked with the double dagger ‡ symbol.
In chemistry, a reactive intermediate or an intermediate is a short-lived, high-energy, highly reactive molecule. When generated in a chemical reaction, it will quickly convert into a more stable molecule. Only in exceptional cases can these compounds be isolated and stored, e.g. low temperatures, matrix isolation. When their existence is indicated, reactive intermediates can help explain how a chemical reaction takes place.
The Brønsted catalysis equation or law of correlation, after Johannes Nicolaus Brønsted, gives the relationship between acid strength and catalytic activity in general acid catalysis.

A nucleophilic aromatic substitution is a substitution reaction in organic chemistry in which the nucleophile displaces a good leaving group, such as a halide, on an aromatic ring. There are 6 nucleophilic substitution mechanisms encountered with aromatic systems:

Hammond's postulate, is a hypothesis in physical organic chemistry which describes the geometric structure of the transition state in an organic chemical reaction. First proposed by George Hammond in 1955, the postulate states that:
If two states, as, for example, a transition state and an unstable intermediate, occur consecutively during a reaction process and have nearly the same energy content, their interconversion will involve only a small reorganization of the molecular structures.
The Hammett equation in organic chemistry describes a linear free-energy relationship relating reaction rates and equilibrium constants for many reactions involving benzoic acid derivatives with meta- and para-substituents to each other with just two parameters: a substituent constant and a reaction constant. This equation was developed and published by Louis Plack Hammett in 1937 as a follow-up to qualitative observations in a 1935 publication.
The alpha effect refers to the increased nucleophilicity of an atom due to the presence of an adjacent (alpha) atom with lone pair electrons. This first atom does not necessarily exhibit increased basicity compared with a similar atom without an adjacent electron donating atom. The effect is well established with many theories to explain the effect but without a clear winner.
Enzyme catalysis is the increase in the rate of a chemical reaction by the active site of a protein. The protein catalyst (enzyme) may be part of a multi-subunit complex, and/or may transiently or permanently associate with a Cofactor. Catalysis of biochemical reactions in the cell is vital due to the very low reaction rates of the uncatalysed reactions at room temperature and pressure. A key driver of protein evolution is the optimization of such catalytic activities via protein dynamics.

Transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.

For a chemical reaction or process an energy profile is a theoretical representation of a single energetic pathway, along the reaction coordinate, as the reactants are transformed into products. Reaction coordinate diagrams are derived from the corresponding potential energy surface (PES), which are used in computational chemistry to model chemical reactions by relating the energy of a molecule(s) to its structure. The reaction coordinate is a parametric curve that follows the pathway of a reaction and indicates the progress of a reaction.
The Taft equation is a linear free energy relationship (LFER) used in physical organic chemistry in the study of reaction mechanisms and in the development of quantitative structure–activity relationships for organic compounds. It was developed by Robert W. Taft in 1952 as a modification to the Hammett equation. While the Hammett equation accounts for how field, inductive, and resonance effects influence reaction rates, the Taft equation also describes the steric effects of a substituent. The Taft equation is written as:
Enthalpy–entropy compensation is a specific example of the compensation effect. The compensation effect refers to the behavior of a series of closely related chemical reactions, which exhibit a linear relationship between one of the following kinetic or thermodynamic parameters for describing the reactions:
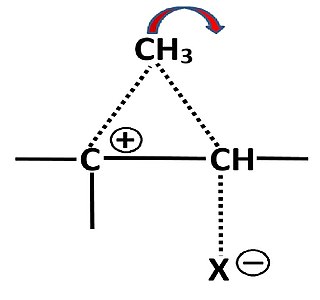
Nonclassical carbocations are stabilized by charge delocalization from contributions of neighbouring C–C or C–H bonds, which can form bridged intermediates or transition states. Nonclassical ions have been extensively studied with the 2-norbornyl system, which as “naked” ion unambiguously exhibit such a bridged structure. The landmark of nonclassical ions are unexpectedly fast solvolysis rates and large differences between epimeric esters. Such behaviour is not restricted to 2-norbornyl esters, as has been shown with some cyclopentyl and steroidal esters with the tosyloxy leaving group.
George Simms Hammond was an American scientist and theoretical chemist who developed "Hammond's postulate", and fathered organic photochemistry,–the general theory of the geometric structure of the transition state in an organic chemical reaction. Hammond's research is also known for its influence on the philosophy of science. His research garnered him the Norris Award in 1968, the Priestley Medal in 1976, the National Medal of Science in 1994, and the Othmer Gold Medal in 2003. He served as the executive chairman of the Allied Chemical Corporation from 1979 to 1989.
In physical organic chemistry, the Grunwald–Winstein equation is a linear free energy relationship between relative rate constants and the ionizing power of various solvent systems, describing the effect of solvent as nucleophile on different substrates. The equation, which was developed by Ernest Grunwald and Saul Winstein in 1948, could be written
In chemistry, solvent effects are the influence of a solvent on chemical reactivity or molecular associations. Solvents can have an effect on solubility, stability and reaction rates and choosing the appropriate solvent allows for thermodynamic and kinetic control over a chemical reaction.