Materials

Phi 29 DNA polymerase
Bacteriophage Φ29 DNA polymerase is a high-processivity enzyme that can produce DNA amplicons greater than 70 kilobase pairs. [1] Its high fidelity and 3'–5' proofreading activity reduces the amplification error rate to 1 in 106−107 bases compared to conventional Taq polymerase with a reported error rate of 1 in 9,000. [2] The reaction can be carried out at a moderate isothermal condition of 30 °C and therefore does not require a thermocycler. It has been actively used in cell-free cloning, which is the enzymatic method of amplifying DNA in vitro without cell culturing and DNA extraction. The large fragment of Bst DNA polymerase is also used in MDA, but Ф29 is generally preferred due to its sufficient product yield and proofreading activity. [3]
Hexamer primers
Hexamer primers are sequences composed of six random nucleotides. For MDA applications, these primers are usually thiophosphate-modified at their 3' end to convey resistance to the 3'–5' exonuclease activity of Ф29 DNA polymerase. MDA reactions start with the annealing of such primers to the DNA template followed by polymerase-mediated chain elongation. Increasing numbers of primer annealing events happen along the amplification reaction.
Reaction
The amplification reaction initiates when multiple primer hexamers anneal to the template. When DNA synthesis proceeds to the next starting site, the polymerase displaces the newly produced DNA strand and continues its strand elongation. The strand displacement generates a newly synthesized single-stranded DNA template for more primers to anneal. Further primer annealing and strand displacement on the newly synthesized template results in a hyper-branched DNA network. The sequence debranching during amplification results in a high yield of the products. To separate the DNA branching network, S1 nucleases are used to cleave the fragments at displacement sites. The nicks on the resulting DNA fragments are repaired by DNA polymerase I.
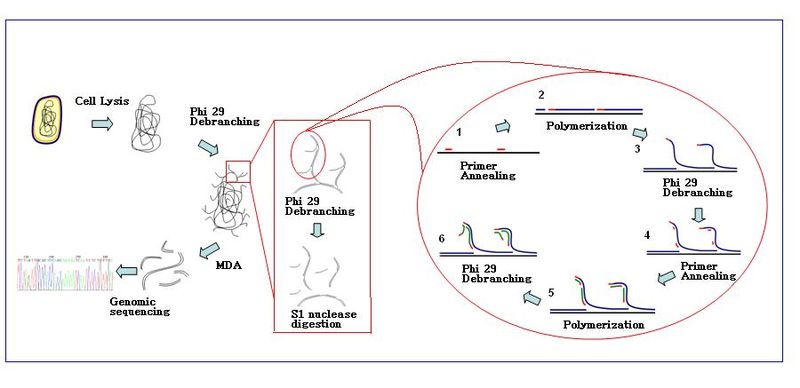
Product quality
MDA can generate 1–2 μg of DNA from single cell with genome coverage of up to 99%. [4] Products also have lower error rate and larger sizes compared to PCR based Taq amplification. [4] [5]
General work flow of MDA: [6]
- Sample preparation: Samples are collected and diluted in the appropriate reaction buffer (Ca2+ and Mg2+ free). Cells are lysed with alkaline buffer.
- Condition: The MDA reaction with Ф29 polymerase is carried out at 30 °C. The reaction usually takes about 2.5–3 hours.
- End of reaction: Inactivate enzymes at 65 °C before collection of the amplified DNA products
- DNA products can be purified with commercial purification kit.