Related Research Articles

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.
Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, the evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.
The coding region of a gene, also known as the coding sequence(CDS), is the portion of a gene's DNA or RNA that codes for a protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.

Alternative splicing, or alternative RNA splicing, or differential splicing, is an alternative splicing process during gene expression that allows a single gene to code for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. This means the exons are joined in different combinations, leading to different (alternative) mRNA strands. Consequently, the proteins translated from alternatively spliced mRNAs usually contain differences in their amino acid sequence and, often, in their biological functions.
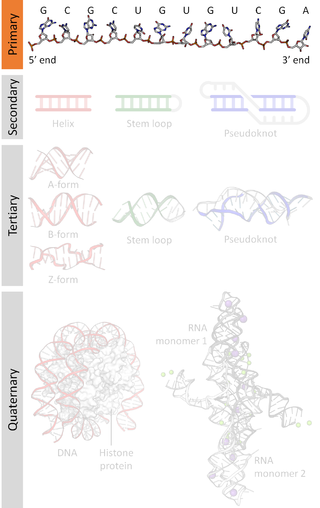
A nucleic acid sequence is a succession of bases within the nucleotides forming alleles within a DNA or RNA (GACU) molecule. This succession is denoted by a series of a set of five different letters that indicate the order of the nucleotides. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, with its double helix, there are two possible directions for the notated sequence; of these two, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.

A frameshift mutation is a genetic mutation caused by indels of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame, resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein. A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.

A point mutation is a genetic mutation where a single nucleotide base is changed, inserted or deleted from a DNA or RNA sequence of an organism's genome. Point mutations have a variety of effects on the downstream protein product—consequences that are moderately predictable based upon the specifics of the mutation. These consequences can range from no effect to deleterious effects, with regard to protein production, composition, and function.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "candidate-gene" approach.
Silent mutations are mutations in DNA that do not have an observable effect on the organism's phenotype. They are a specific type of neutral mutation. The phrase silent mutation is often used interchangeably with the phrase synonymous mutation; however, synonymous mutations are not always silent, nor vice versa. Synonymous mutations can affect transcription, splicing, mRNA transport, and translation, any of which could alter phenotype, rendering the synonymous mutation non-silent. The substrate specificity of the tRNA to the rare codon can affect the timing of translation, and in turn the co-translational folding of the protein. This is reflected in the codon usage bias that is observed in many species. Mutations that cause the altered codon to produce an amino acid with similar functionality are often classified as silent; if the properties of the amino acid are conserved, this mutation does not usually significantly affect protein function.
Genetics, a discipline of biology, is the science of heredity and variation in living organisms.

In genetics, an insertion is the addition of one or more nucleotide base pairs into a DNA sequence. This can often happen in microsatellite regions due to the DNA polymerase slipping. Insertions can be anywhere in size from one base pair incorrectly inserted into a DNA sequence to a section of one chromosome inserted into another. The mechanism of the smallest single base insertion mutations is believed to be through base-pair separation between the template and primer strands followed by non-neighbor base stacking, which can occur locally within the DNA polymerase active site. On a chromosome level, an insertion refers to the insertion of a larger sequence into a chromosome. This can happen due to unequal crossover during meiosis.

In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.

A gene is said to be polymorphic if more than one allele occupies that gene's locus within a population. In addition to having more than one allele at a specific locus, each allele must also occur in the population at a rate of at least 1% to generally be considered polymorphic.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.
References
- ↑ Schwarz, Jana Marie; Rödelsperger, Christian; Schuelke, Markus; Seelow, Dominik (2010-08-01). "MutationTaster evaluates disease-causing potential of sequence alterations". Nature Methods. 7 (8): 575–576. doi:10.1038/nmeth0810-575. ISSN 1548-7105. PMID 20676075. S2CID 26892938.
- ↑ Schwarz, Jana Marie; Cooper, David N; Schuelke, Markus; Seelow, Dominik (2014-03-28). "MutationTaster2: mutation prediction for the deep-sequencing age". Nature Methods. 11 (4): 361–362. doi:10.1038/nmeth.2890. ISSN 1548-7105. PMID 24681721. S2CID 19382079.
- ↑ Wheeler, David A. (2008-04-17). "The complete genome of an individual by massively parallel DNA sequencing". Nature. 452 (7189): 872–876. Bibcode:2008Natur.452..872W. doi: 10.1038/nature06884 . PMID 18421352.
- ↑ Simcikova D, Heneberg P (December 2019). "Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases". Scientific Reports. 9 (1): 18577. Bibcode:2019NatSR...918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466 . PMID 31819097.