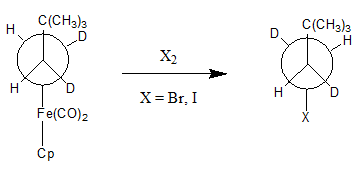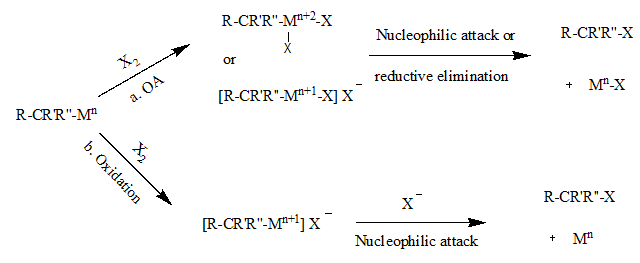In chemistry, an acyl group is a moiety derived by the removal of one or more hydroxyl groups from an oxoacid, including inorganic acids. It contains a double-bonded oxygen atom and an organyl group or hydrogen in the case of formyl group. In organic chemistry, the acyl group is usually derived from a carboxylic acid, in which case it has the formula R−C(=O)−, where R represents an organyl group or hydrogen. Although the term is almost always applied to organic compounds, acyl groups can in principle be derived from other types of acids such as sulfonic acids and phosphonic acids. In the most common arrangement, acyl groups are attached to a larger molecular fragment, in which case the carbon and oxygen atoms are linked by a double bond.

An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism. The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism.

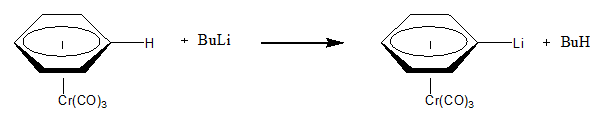
In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
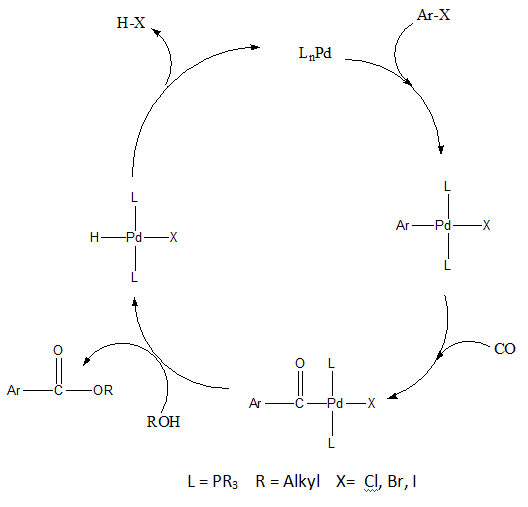
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
In organic chemistry, the diazo group is an organic moiety consisting of two linked nitrogen atoms at the terminal position. Overall charge-neutral organic compounds containing the diazo group bound to a carbon atom are called diazo compounds or diazoalkanes and are described by the general structural formula R2C=N+=N−. The simplest example of a diazo compound is diazomethane, CH2N2. Diazo compounds should not be confused with azo compounds or with diazonium compounds.
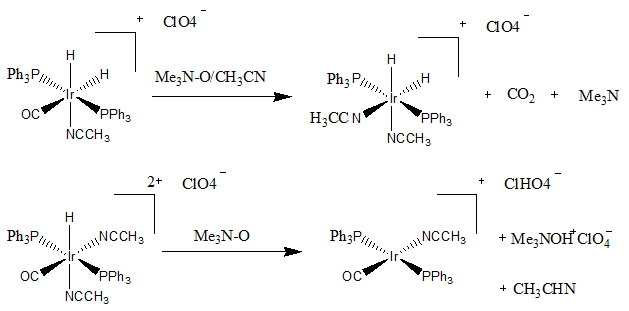
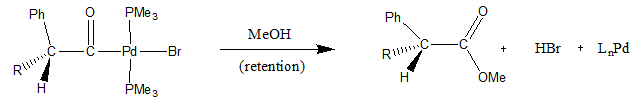
Oxidative addition and reductive elimination are two important and related classes of reactions in organometallic chemistry. Oxidative addition is a process that increases both the oxidation state and coordination number of a metal centre. Oxidative addition is often a step in catalytic cycles, in conjunction with its reverse reaction, reductive elimination.

Organoboron chemistry or organoborane chemistry studies organoboron compounds, also called organoboranes. These chemical compounds combine boron and carbon; typically, they are organic derivatives of borane (BH3), as in the trialkyl boranes.
A transition metal carbene complex is an organometallic compound featuring a divalent organic ligand. The divalent organic ligand coordinated to the metal center is called a carbene. Carbene complexes for almost all transition metals have been reported. Many methods for synthesizing them and reactions utilizing them have been reported. The term carbene ligand is a formalism since many are not derived from carbenes and almost none exhibit the reactivity characteristic of carbenes. Described often as M=CR2, they represent a class of organic ligands intermediate between alkyls (−CR3) and carbynes (≡CR). They feature in some catalytic reactions, especially alkene metathesis, and are of value in the preparation of some fine chemicals.
Organophosphorus chemistry is the scientific study of the synthesis and properties of organophosphorus compounds, which are organic compounds containing phosphorus. They are used primarily in pest control as an alternative to chlorinated hydrocarbons that persist in the environment. Some organophosphorus compounds are highly effective insecticides, although some are extremely toxic to humans, including sarin and VX nerve agents.

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.

In organometallic chemistry, a metallacycle is a derivative of a carbocyclic compound wherein a metal has replaced at least one carbon center; this is to some extent similar to heterocycles. Metallacycles appear frequently as reactive intermediates in catalysis, e.g. olefin metathesis and alkyne trimerization. In organic synthesis, directed ortho metalation is widely used for the functionalization of arene rings via C-H activation. One main effect that metallic atom substitution on a cyclic carbon compound is distorting the geometry due to the large size of typical metals.
In organometallic chemistry, a migratory insertion is a type of reaction wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
Organoiron chemistry is the chemistry of iron compounds containing a carbon-to-iron chemical bond. Organoiron compounds are relevant in organic synthesis as reagents such as iron pentacarbonyl, diiron nonacarbonyl and disodium tetracarbonylferrate. While iron adopts oxidation states from Fe(−II) through to Fe(VII), Fe(IV) is the highest established oxidation state for organoiron species. Although iron is generally less active in many catalytic applications, it is less expensive and "greener" than other metals. Organoiron compounds feature a wide range of ligands that support the Fe-C bond; as with other organometals, these supporting ligands prominently include phosphines, carbon monoxide, and cyclopentadienyl, but hard ligands such as amines are employed as well.

Organoruthenium chemistry is the chemistry of organometallic compounds containing a carbon to ruthenium chemical bond. Several organoruthenium catalysts are of commercial interest and organoruthenium compounds have been considered for cancer therapy. The chemistry has some stoichiometric similarities with organoiron chemistry, as iron is directly above ruthenium in group 8 of the periodic table. The most important reagents for the introduction of ruthenium are ruthenium(III) chloride and triruthenium dodecacarbonyl.

Organomolybdenum chemistry is the chemistry of chemical compounds with Mo-C bonds. The heavier group 6 elements molybdenum and tungsten form organometallic compounds similar to those in organochromium chemistry but higher oxidation states tend to be more common.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
In chemistry, mesoionic carbenes (MICs) are a type of reactive intermediate that are related to N-heterocyclic carbenes (NHCs); thus, MICs are also referred to as abnormal N-heterocyclic carbenes (aNHCs) or remote N-heterocyclic carbenes (rNHCs). Unlike simple NHCs, the canonical resonance structures of these carbenes are mesoionic: an MIC cannot be drawn without adding additional charges to some of the atoms.
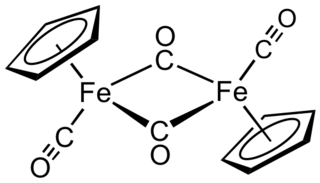
Cyclopentadienyliron dicarbonyl dimer is an organometallic compound with the formula [(η5-C5H5)Fe(CO)2]2, often abbreviated to Cp2Fe2(CO)4, [CpFe(CO)2]2 or even Fp2, with the colloquial name "fip dimer". It is a dark reddish-purple crystalline solid, which is readily soluble in moderately polar organic solvents such as chloroform and pyridine, but less soluble in carbon tetrachloride and carbon disulfide. Cp2Fe2(CO)4 is insoluble in but stable toward water. Cp2Fe2(CO)4 is reasonably stable to storage under air and serves as a convenient starting material for accessing other Fp (CpFe(CO)2) derivatives (described below).
A metal carbido complex is a coordination complex that contains a carbon atom as a ligand. They are analogous to metal nitrido complexes. Carbido complexes are a molecular subclass of carbides, which are prevalent in organometallic and inorganic chemistry. Carbido complexes represent models for intermediates in Fischer–Tropsch synthesis, olefin metathesis, and related catalytic industrial processes. Ruthenium-based carbido complexes are by far the most synthesized and characterized to date. Although, complexes containing chromium, gold, iron, nickel, molybdenum, osmium, rhenium, and tungsten cores are also known. Mixed-metal carbides are also known.
A Fischer carbene is a type of transition metal carbene complex, which is an organometallic compound containing a divalent organic ligand. In a Fischer carbene, the carbene ligand is a σ-donor π-acceptor ligand. Because π-backdonation from the metal centre is generally weak, the carbene carbon is electrophilic.