
In stereochemistry, stereoisomerism, or spatial isomerism, is a form of isomerism in which molecules have the same molecular formula and sequence of bonded atoms (constitution), but differ in the three-dimensional orientations of their atoms in space. This contrasts with structural isomers, which share the same molecular formula, but the bond connections or their order differs. By definition, molecules that are stereoisomers of each other represent the same structural isomer.
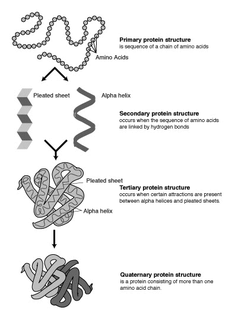
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

A dihedral angle is the angle between two intersecting planes or half-planes. In chemistry, it is the clockwise angle between half-planes through two sets of three atoms, having two atoms in common. In solid geometry, it is defined as the union of a line and two half-planes that have this line as a common edge. In higher dimensions, a dihedral angle represents the angle between two hyperplanes. The planes of a flying machine are said to be at positive dihedral angle when both starboard and port main planes are upwardly inclined to the lateral axis. When downwardly inclined they are said to be at a negative dihedral angle.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
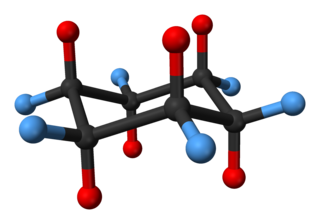
In organic chemistry, cyclohexane conformations are any of several three-dimensional shapes adopted by molecules of cyclohexane. Because many compounds feature structurally similar six-membered rings, the structure and dynamics of cyclohexane are important prototypes of a wide range of compounds.

In chemistry, conformational isomerism is a form of stereoisomerism in which the isomers can be interconverted just by rotations about formally single bonds. While any two arrangements of atoms in a molecule that differ by rotation about single bonds can be referred to as different conformations, conformations that correspond to local minima on the potential energy surface are specifically called conformational isomers or conformers. Conformations that correspond to local maxima on the energy surface are the transition states between the local-minimum conformational isomers. Rotations about single bonds involve overcoming a rotational energy barrier to interconvert one conformer to another. If the energy barrier is low, there is free rotation and a sample of the compound exists as a rapidly equilibrating mixture of multiple conformers; if the energy barrier is high enough then there is restricted rotation, a molecule may exist for a relatively long time period as a stable rotational isomer or rotamer. When the time scale for interconversion is long enough for isolation of individual rotamers, the isomers are termed atropisomers. The ring-flip of substituted cyclohexanes constitutes another common form of conformational isomerism.
In chemistry, a molecule experiences strain when its chemical structure undergoes some stress which raises its internal energy in comparison to a strain-free reference compound. The internal energy of a molecule consists of all the energy stored within it. A strained molecule has an additional amount of internal energy which an unstrained molecule does not. This extra internal energy, or strain energy, can be likened to a compressed spring. Much like a compressed spring must be held in place to prevent release of its potential energy, a molecule can be held in an energetically unfavorable conformation by the bonds within that molecule. Without the bonds holding the conformation in place, the strain energy would be released.

In organic chemistry, the anomeric effect or Edward-Lemieux effect is a stereoelectronic effect that describes the tendency of heteroatomic substituents adjacent to a heteroatom within a cyclohexane ring to prefer the axial orientation instead of the less hindered equatorial orientation that would be expected from steric considerations. This effect was originally observed in pyranose rings by J. T. Edward in 1955 when studying carbohydrate chemistry.

In the study of conformational isomerism, the Gauche effect is an atypical situation where a gauche conformation is more stable than the anti conformation (180°).

Allylic strain in organic chemistry is a type of strain energy resulting from the interaction between a substituent on one end of an olefin with an allylic substituent on the other end. If the substituents are large enough in size, they can sterically interfere with each other such that one conformer is greatly favored over the other. Allylic strain was first recognized in the literature in 1965 by Johnson and Malhotra. The authors were investigating cyclohexane conformations including endocyclic and exocylic double bonds when they noticed certain conformations were disfavored due to the geometry constraints caused by the double bond. Organic chemists capitalize on the rigidity resulting from allylic strain for use in asymmetric reactions.

The carbon–fluorine bond is a polar covalent bond between carbon and fluorine that is a component of all organofluorine compounds. It is one of the strongest single bonds in chemistry, and relatively short, due to its partial ionic character. The bond also strengthens and shortens as more fluorines are added to the same carbon on a chemical compound. As such, fluoroalkanes like tetrafluoromethane are some of the most unreactive organic compounds.
Methylcyclohexane (cyclohexylmethane) is an organic compound with the molecular formula is CH3C6H11. Classified as saturated hydrocarbon, it is a colourless liquid with a faint odor. Methylcyclohexane is used as a solvent. It is mainly converted in naphtha reformers to toluene. Methylcyclohexane is also used in some correction fluids (such as White-Out) as a solvent.
A-Values are numerical values used in the determination of the most stable orientation of atoms in a molecule, as well as a general representation of steric bulk. A-values are derived from energy measurements of the different cyclohexane conformations of a monosubstituted cyclohexane chemical. Substituents on a cyclohexane ring prefer to reside in the equatorial position to the axial. The difference in Gibbs free energy (ΔG) between the higher energy conformation and the lower energy conformation is the A-value for that particular substituent.
In chemistry, isomers are molecules or polyatomic ions with identical molecular formulae – that is, same number of atoms of each element – but distinct arrangements of atoms in space. Isomerism is existence or possibility of isomers.
Carbohydrate conformation refers to the overall three-dimensional structure adopted by a carbohydrate (saccharide) molecule as a result of the through-bond and through-space physical forces it experiences arising from its molecular structure. The physical forces that dictate the three-dimensional shapes of all molecules—here, of all monosaccharide, oligosaccharide, and polysaccharide molecules—are sometimes summarily captured by such terms as "steric interactions" and "stereoelectronic effects".
FoldX is a protein design algorithm that uses an empirical force field. It can determine the energetic effect of point mutations as well as the interaction energy of protein complexes. FoldX can mutate protein and DNA side chains using a probability-based rotamer library, while exploring alternative conformations of the surrounding side chains.
In stereochemistry, the Klyne–Prelog system for describing conformations about a single bond offers a more systematic means to unambiguously name complex structures, where the torsional or dihedral angles are not found to occur in 60° increments. Klyne notation views the placement of the substituent on the front atom as being in regions of space called anti/syn and clinal/periplanar relative to a reference group on the rear atom. A plus (+) or minus (–) sign is placed at the front to indicate the sign of the dihedral angle. Anti or syn indicates the substituents are on opposite sides or the same side, respectively. Clinal substituents are found within 30° of either side of a dihedral angle of 60°, 120° (90°–150°), 240° (210°–270°), or 300° (270°–330°). Periplanar substituents are found within 30° of either 0° (330°–30°) or 180° (150°–210°). Juxtaposing the designations produces the following terms for the conformers of butane : gauche butane is syn-clinal, anti butane is anti-periplanar, and eclipsed butane is syn-periplanar.
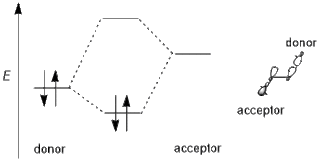
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals. Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap. Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons in space.
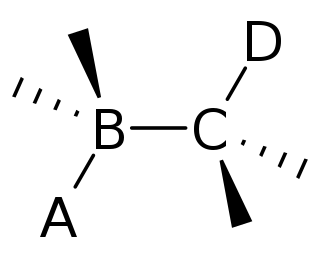
In organic chemistry, anti-periplanar, or antiperiplanar, describes the A−B−C−D bond angle in a molecule. In this conformer, the dihedral angle of the A−B bond and the C−D bond is greater than +150° or less than −150°. Anti-periplanar is often used in textbooks to mean strictly anti-coplanar, with an A−BC−D dihedral angle of 180°. In a Newman projection, the molecule will be in a staggered arrangement with the anti-periplanar functional groups pointing up and down, 180° away from each other. Figure 5 shows 2-chloro-2,3-dimethylbutane in a sawhorse projection with chlorine and a hydrogen anti-periplanar to each other.

In biochemistry, a backbone-dependent rotamer library provides the frequencies, mean dihedral angles, and standard deviations of the discrete conformations of the amino acid side chains in proteins as a function of the backbone dihedral angles φ and ψ of the Ramachandran map. By contrast, backbone-independent rotamer libraries express the frequencies and mean dihedral angles for all side chains in proteins, regardless of the backbone conformation of each residue type. Backbone-dependent rotamer libraries have been shown to have significant advantages over backbone-independent rotamer libraries, principally when used as an energy term, by speeding up search times of side-chain packing algorithms used in protein structure prediction and protein design.

