Related Research Articles

In genetics, a promoter is a sequence of DNA to which proteins bind that initiate transcription of a single RNA from the DNA downstream of it. This RNA may encode a protein, or can have a function in and of itself, such as tRNA, mRNA, or rRNA. Promoters are located near the transcription start sites of genes, upstream on the DNA . Promoters can be about 100–1000 base pairs long, the sequence of which is highly dependent on the gene and product of transcription, type or class of RNA polymerase recruited to the site and species of organism.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
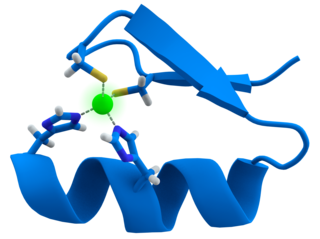
A zinc finger is a small protein structural motif that is characterized by the coordination of one or more zinc ions (Zn2+) in order to stabilize the fold. It was originally coined to describe the finger-like appearance of a hypothesized structure from the African clawed frog (Xenopus laevis) transcription factor IIIA. However, it has been found to encompass a wide variety of differing protein structures in eukaryotic cells. Xenopus laevis TFIIIA was originally demonstrated to contain zinc and require the metal for function in 1983, the first such reported zinc requirement for a gene regulatory protein. It often appears as a metal-binding domain in multi-domain proteins.
In biology, a sequence motif is a nucleotide or amino-acid sequence pattern that is widespread and usually assumed to be related to biological function of the macromolecule. For example, an N-glycosylation site motif can be defined as Asn, followed by anything but Pro, followed by either Ser or Thr, followed by anything but Pro residue.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.
In molecular biology, the TATA box is a sequence of DNA found in the core promoter region of genes in archaea and eukaryotes. The bacterial homolog of the TATA box is called the Pribnow box which has a shorter consensus sequence.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.

In bioinformatics, a sequence logo is a graphical representation of the sequence conservation of nucleotides or amino acids . A sequence logo is created from a collection of aligned sequences and depicts the consensus sequence and diversity of the sequences. Sequence logos are frequently used to depict sequence characteristics such as protein-binding sites in DNA or functional units in proteins.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional “gene-by-gene” approach.

A regulator gene, regulator, or regulatory gene is a gene involved in controlling the expression of one or more other genes. Regulatory sequences, which encode regulatory genes, are often at the five prime end (5') to the start site of transcription of the gene they regulate. In addition, these sequences can also be found at the three prime end (3') to the transcription start site. In both cases, whether the regulatory sequence occurs before (5') or after (3') the gene it regulates, the sequence is often many kilobases away from the transcription start site. A regulator gene may encode a protein, or it may work at the level of RNA, as in the case of genes encoding microRNAs. An example of a regulator gene is a gene that codes for a repressor protein that inhibits the activity of an operator.
A position weight matrix (PWM), also known as a position-specific weight matrix (PSWM) or position-specific scoring matrix (PSSM), is a commonly used representation of motifs (patterns) in biological sequences.
Cis-regulatory elements (CREs) or Cis-regulatory modules (CRMs) are regions of non-coding DNA which regulate the transcription of neighboring genes. CREs are vital components of genetic regulatory networks, which in turn control morphogenesis, the development of anatomy, and other aspects of embryonic development, studied in evolutionary developmental biology.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.
DNA binding sites are a type of binding site found in DNA where other molecules may bind. DNA binding sites are distinct from other binding sites in that (1) they are part of a DNA sequence and (2) they are bound by DNA-binding proteins. DNA binding sites are often associated with specialized proteins known as transcription factors, and are thus linked to transcriptional regulation. The sum of DNA binding sites of a specific transcription factor is referred to as its cistrome. DNA binding sites also encompasses the targets of other proteins, like restriction enzymes, site-specific recombinases and methyltransferases.
Protein function prediction methods are techniques that bioinformatics researchers use to assign biological or biochemical roles to proteins. These proteins are usually ones that are poorly studied or predicted based on genomic sequence data. These predictions are often driven by data-intensive computational procedures. Information may come from nucleic acid sequence homology, gene expression profiles, protein domain structures, text mining of publications, phylogenetic profiles, phenotypic profiles, and protein-protein interaction. Protein function is a broad term: the roles of proteins range from catalysis of biochemical reactions to transport to signal transduction, and a single protein may play a role in multiple processes or cellular pathways.
TRANSFAC is a manually curated database of eukaryotic transcription factors, their genomic binding sites and DNA binding profiles. The contents of the database can be used to predict potential transcription factor binding sites.

The WRKY domain is found in the WRKY transcription factor family, a class of transcription factors. The WRKY domain is found almost exclusively in plants although WRKY genes appear present in some diplomonads, social amoebae and other amoebozoa, and fungi incertae sedis. They appear absent in other non-plant species. WRKY transcription factors have been a significant area of plant research for the past 20 years. The WRKY DNA-binding domain recognizes the W-box (T)TGAC(C/T) cis-regulatory element.
CUT&RUN-sequencing, also known as cleavage under targets and release using nuclease, is a method used to analyze protein interactions with DNA. CUT&RUN-sequencing combines antibody-targeted controlled cleavage by micrococcal nuclease with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN-sequencing does not.
References
- ↑ Palumbo, MJ; Newberg, LA (July 1, 2010). "Phyloscan: locating transcription-regulating binding sites in mixed aligned and unaligned sequence data". Nucleic Acids Research. 38 (Web server issue): W268–W274. doi:10.1093/nar/gkq330. PMC 2896078 . PMID 20435683.
- ↑ Carmack, CS; McCue, LA; Newberg, LA; Lawrence, CE (January 23, 2007). "PhyloScan: identification of transcription factor binding sites using cross-species evidence". Algorithms for Molecular Biology. 2 (1): article 1. doi:10.1186/1748-7188-2-1. PMC 1794230 . PMID 17244358.
- ↑ Staden, R (April 1989). "Methods for calculating the probabilities of finding patterns in sequences". Computer Applications in the Biosciences. 5 (2): 89–96. doi:10.1093/bioinformatics/5.2.89. PMID 2720468.
- ↑ Moses, AM; Chiang, DY; Pollard, DA; Iyer, VN; Eisen, MB (November 30, 2004). "MONKEY: identifying conserved transcription-factor binding sites in multiple alignments using a binding site-specific evolutionary model". Genome Biology. 5 (12): R98. doi:10.1186/gb-2004-5-12-r98. PMC 545801 . PMID 15575972.
- ↑ Bailey, TL; Gribskov, M (Summer 1998). "Methods and statistics for combining motif match scores". Journal of Computational Biology. 5 (2): 211–221. doi:10.1089/cmb.1998.5.211. PMID 9672829.
- ↑ Neuwald, AF; Green, P (June 24, 1994). "Detecting patterns in protein sequences". Journal of Molecular Biology. 239 (5): 698–712. doi:10.1006/jmbi.1994.1407. PMID 8014990.