Computational chemistry is a branch of chemistry that uses computer simulation to assist in solving chemical problems. It uses methods of theoretical chemistry, incorporated into computer programs, to calculate the structures and properties of molecules, groups of molecules, and solids. It is essential because, apart from relatively recent results concerning the hydrogen molecular ion, the quantum many-body problem cannot be solved analytically, much less in closed form. While computational results normally complement the information obtained by chemical experiments, it can in some cases predict hitherto unobserved chemical phenomena. It is widely used in the design of new drugs and materials.

Sir John Anthony Pople was a British theoretical chemist who was awarded the Nobel Prize in Chemistry with Walter Kohn in 1998 for his development of computational methods in quantum chemistry.
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.
A linear combination of atomic orbitals or LCAO is a quantum superposition of atomic orbitals and a technique for calculating molecular orbitals in quantum chemistry. In quantum mechanics, electron configurations of atoms are described as wavefunctions. In a mathematical sense, these wave functions are the basis set of functions, the basis functions, which describe the electrons of a given atom. In chemical reactions, orbital wavefunctions are modified, i.e. the electron cloud shape is changed, according to the type of atoms participating in the chemical bond.
In computational physics and chemistry, the Hartree–Fock (HF) method is a method of approximation for the determination of the wave function and the energy of a quantum many-body system in a stationary state.
In chemistry, molecular orbital theory is a method for describing the electronic structure of molecules using quantum mechanics. It was proposed early in the 20th century.

MOLPRO is a software package used for accurate ab initio quantum chemistry calculations. It is developed by Peter Knowles at Cardiff University and Hans-Joachim Werner at Universität Stuttgart in collaboration with other authors.
Gaussian is a general purpose computational chemistry software package initially released in 1970 by John Pople and his research group at Carnegie Mellon University as Gaussian 70. It has been continuously updated since then. The name originates from Pople's use of Gaussian orbitals to speed up molecular electronic structure calculations as opposed to using Slater-type orbitals, a choice made to improve performance on the limited computing capacities of then-current computer hardware for Hartree–Fock calculations. The current version of the program is Gaussian 16. Originally available through the Quantum Chemistry Program Exchange, it was later licensed out of Carnegie Mellon University, and since 1987 has been developed and licensed by Gaussian, Inc.
Q-Chem is a general-purpose electronic structure package featuring a variety of established and new methods implemented using innovative algorithms that enable fast calculations of large systems on various computer architectures, from laptops and regular lab workstations to midsize clusters and HPCC, using density functional and wave-function based approaches. It offers an integrated graphical interface and input generator; a large selection of functionals and correlation methods, including methods for electronically excited states and open-shell systems; solvation models; and wave-function analysis tools. In addition to serving the computational chemistry community, Q-Chem also provides a versatile code development platform.
Møller–Plesset perturbation theory (MP) is one of several quantum chemistry post–Hartree–Fock ab initio methods in the field of computational chemistry. It improves on the Hartree–Fock method by adding electron correlation effects by means of Rayleigh–Schrödinger perturbation theory (RS-PT), usually to second (MP2), third (MP3) or fourth (MP4) order. Its main idea was published as early as 1934 by Christian Møller and Milton S. Plesset.
Electronic correlation is the interaction between electrons in the electronic structure of a quantum system. The correlation energy is a measure of how much the movement of one electron is influenced by the presence of all other electrons.
In computational chemistry, post-Hartree–Fock methods are the set of methods developed to improve on the Hartree–Fock (HF), or self-consistent field (SCF) method. They add electron correlation which is a more accurate way of including the repulsions between electrons than in the Hartree–Fock method where repulsions are only averaged.
In theoretical and computational chemistry, a basis set is a set of functions that is used to represent the electronic wave function in the Hartree–Fock method or density-functional theory in order to turn the partial differential equations of the model into algebraic equations suitable for efficient implementation on a computer.
CNDO is the abbreviation for Complete Neglect of Differential Overlap, one of the first semi empirical methods in quantum chemistry. It uses two approximations:

PQS is a general purpose quantum chemistry program. Its roots go back to the first ab initio gradient program developed in Professor Peter Pulay's group but now it is developed and distributed commercially by Parallel Quantum Solutions. There is a reduction in cost for academic users and a site license. Its strong points are geometry optimization, NMR chemical shift calculations, and large MP2 calculations, and high parallel efficiency on computing clusters. It includes many other capabilities including Density functional theory, the semiempirical methods, MINDO/3, MNDO, AM1 and PM3, Molecular mechanics using the SYBYL 5.0 Force Field, the quantum mechanics/molecular mechanics mixed method using the ONIOM method, natural bond orbital (NBO) analysis and COSMO solvation models. Recently, a highly efficient parallel CCSD(T) code for closed shell systems has been developed. This code includes many other post Hartree–Fock methods: MP2, MP3, MP4, CISD, CEPA, QCISD and so on.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Semi-empirical quantum chemistry methods are based on the Hartree–Fock formalism, but make many approximations and obtain some parameters from empirical data. They are very important in computational chemistry for treating large molecules where the full Hartree–Fock method without the approximations is too expensive. The use of empirical parameters appears to allow some inclusion of electron correlation effects into the methods.
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initio was first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene. The background is described by Parr. Ab initio means "from first principles" or "from the beginning", implying that the only inputs into an ab initio calculation are physical constants. Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order to yield useful information such as electron densities, energies and other properties of the system. The ability to run these calculations has enabled theoretical chemists to solve a range of problems and their importance is highlighted by the awarding of the Nobel prize to John Pople and Walter Kohn.
Quantum chemistry composite methods are computational chemistry methods that aim for high accuracy by combining the results of several calculations. They combine methods with a high level of theory and a small basis set with methods that employ lower levels of theory with larger basis sets. They are commonly used to calculate thermodynamic quantities such as enthalpies of formation, atomization energies, ionization energies and electron affinities. They aim for chemical accuracy which is usually defined as within 1 kcal/mol of the experimental value. The first systematic model chemistry of this type with broad applicability was called Gaussian-1 (G1) introduced by John Pople. This was quickly replaced by the Gaussian-2 (G2) which has been used extensively. The Gaussian-3 (G3) was introduced later.
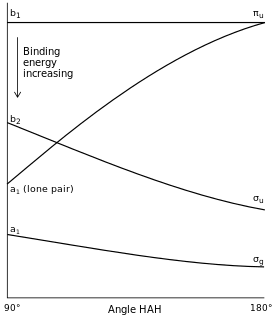
Walsh diagrams, often called angular coordinate diagrams or correlation diagrams, are representations of calculated orbital binding energies of a molecule versus a distortion coordinate, used for making quick predictions about the geometries of small molecules. By plotting the change in molecular orbital levels of a molecule as a function of geometrical change, Walsh diagrams explain why molecules are more stable in certain spatial configurations.

