In chemistry, a nucleophilic substitution is a class of chemical reactions in which an electron-rich chemical species replaces a functional group within another electron-deficient molecule. The molecule that contains the electrophile and the leaving functional group is called the substrate.

An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism. The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism.
The SN1 reaction is a substitution reaction in organic chemistry, the name of which refers to the Hughes-Ingold symbol of the mechanism. "SN" stands for "nucleophilic substitution", and the "1" says that the rate-determining step is unimolecular. Thus, the rate equation is often shown as having first-order dependence on the substrate and zero-order dependence on the nucleophile. This relationship holds for situations where the amount of nucleophile is much greater than that of the intermediate. Instead, the rate equation may be more accurately described using steady-state kinetics. The reaction involves a carbocation intermediate and is commonly seen in reactions of secondary or tertiary alkyl halides under strongly basic conditions or, under strongly acidic conditions, with secondary or tertiary alcohols. With primary and secondary alkyl halides, the alternative SN2 reaction occurs. In inorganic chemistry, the SN1 reaction is often known as the dissociative substitution. This dissociation pathway is well-described by the cis effect. A reaction mechanism was first proposed by Christopher Ingold et al. in 1940. This reaction does not depend much on the strength of the nucleophile, unlike the SN2 mechanism. This type of mechanism involves two steps. The first step is the ionization of alkyl halide in the presence of aqueous acetone or ethyl alcohol. This step provides a carbocation as an intermediate.
In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair. Because electrophiles accept electrons, they are Lewis acids. Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
A substitution reaction is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group. Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilic or nucleophilic depending upon the reagent involved, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical, and whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
In chemical kinetics, the overall rate of a reaction is often approximately determined by the slowest step, known as the rate-determining step or rate-limiting step. For a given reaction mechanism, the prediction of the corresponding rate equation is often simplified by using this approximation of the rate-determining step.
In chemical kinetics, a reaction rate constant or reaction rate coefficient is a proportionality constant which quantifies the rate and direction of a chemical reaction by relating it with the concentration of reactants.
A 1,2-rearrangement or 1,2-migration or 1,2-shift or Whitmore 1,2-shift is an organic reaction where a substituent moves from one atom to another atom in a chemical compound. In a 1,2 shift the movement involves two adjacent atoms but moves over larger distances are possible. In the example below the substituent R moves from carbon atom C2 to C3.
In chemistry, molecularity is the number of molecules that come together to react in an elementary (single-step) reaction and is equal to the sum of stoichiometric coefficients of reactants in the elementary reaction with effective collision and correct orientation. Depending on how many molecules come together, a reaction can be unimolecular, bimolecular or even trimolecular.
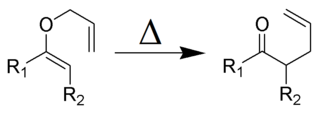
The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation.
The Dowd–Beckwith ring-expansion reaction is an organic reaction in which a cyclic carbonyl is expanded by up to 4 carbons in a free radical ring expansion reaction through an α-alkylhalo substituent. The radical initiator system is based on AIBN and tributyltin hydride. The cyclic β-keto ester can be obtained through a Dieckmann condensation. The original reaction consisted of a nucleophilic aliphatic substitution of the enolate of ethyl cyclohexanone-2-carboxylate with 1,4-diiodobutane and sodium hydride followed by ring expansion to ethyl cyclodecanone-6-carboxylate. A side-reaction is organic reduction of the iodoalkane.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The vinylcyclopropane rearrangement or vinylcyclopropane-cyclopentene rearrangement is a ring expansion reaction, converting a vinyl-substituted cyclopropane ring into a cyclopentene ring.

The vinyl cation is a carbocation with the positive charge on an alkene carbon. Its empirical formula is C
2H+
3. More generally, a vinylic cation is any disubstituted carbon, where the carbon bearing the positive charge is part of a double bond and is sp hybridized. In the chemical literature, substituted vinylic cations are often referred to as vinyl cations, and understood to refer to the broad class rather than the C
2H+
3 variant alone. The vinyl cation is one of the main types of reactive intermediates involving a non-tetrahedrally coordinated carbon atom, and is necessary to explain a wide variety of observed reactivity trends. Vinyl cations are observed as reactive intermediates in solvolysis reactions, as well during electrophilic addition to alkynes, for example, through protonation of an alkyne by a strong acid. As expected from its sp hybridization, the vinyl cation prefers a linear geometry. Compounds related to the vinyl cation include allylic carbocations and benzylic carbocations, as well as aryl carbocations.
In physical organic chemistry, the Grunwald–Winstein equation is a linear free energy relationship between relative rate constants and the ionizing power of various solvent systems, describing the effect of solvent as nucleophile on different substrates. The equation, which was developed by Ernest Grunwald and Saul Winstein in 1948, could be written
Radical cyclization reactions are organic chemical transformations that yield cyclic products through radical intermediates. They usually proceed in three basic steps: selective radical generation, radical cyclization, and conversion of the cyclized radical to product.
In organic chemistry, enone–alkene cycloadditions are a version of the [2+2] cycloaddition This reaction involves an enone and alkene as substrates. Although the concerted photochemical [2+2] cycloaddition is allowed, the reaction between enones and alkenes is stepwise and involves discrete diradical intermediates.




