Bacterial conjugation is the transfer of genetic material between bacterial cells by direct cell-to-cell contact or by a bridge-like connection between two cells. This takes place through a pilus. It is a parasexual mode of reproduction in bacteria.
A bacterial artificial chromosome (BAC) is a DNA construct, based on a functional fertility plasmid, used for transforming and cloning in bacteria, usually E. coli. F-plasmids play a crucial role because they contain partition genes that promote the even distribution of plasmids after bacterial cell division. The bacterial artificial chromosome's usual insert size is 150–350 kbp. A similar cloning vector called a PAC has also been produced from the DNA of P1 bacteriophage.

Chromosomal crossover, or crossing over, is the exchange of genetic material during sexual reproduction between two homologous chromosomes' non-sister chromatids that results in recombinant chromosomes. It is one of the final phases of genetic recombination, which occurs in the pachytene stage of prophase I of meiosis during a process called synapsis. Synapsis begins before the synaptonemal complex develops and is not completed until near the end of prophase I. Crossover usually occurs when matching regions on matching chromosomes break and then reconnect to the other chromosome.

Genetic recombination is the exchange of genetic material between different organisms which leads to production of offspring with combinations of traits that differ from those found in either parent. In eukaryotes, genetic recombination during meiosis can lead to a novel set of genetic information that can be passed on from the parents to the offspring. Most recombination is naturally occurring.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.

An expression vector, otherwise known as an expression construct, is usually a plasmid or virus designed for gene expression in cells. The vector is used to introduce a specific gene into a target cell, and can commandeer the cell's mechanism for protein synthesis to produce the protein encoded by the gene. Expression vectors are the basic tools in biotechnology for the production of proteins.

In molecular biology and genetics, transformation is the genetic alteration of a cell resulting from the direct uptake and incorporation of exogenous genetic material from its surroundings through the cell membrane(s). For transformation to take place, the recipient bacterium must be in a state of competence, which might occur in nature as a time-limited response to environmental conditions such as starvation and cell density, and may also be induced in a laboratory.

RecBCD is an enzyme of the E. coli bacterium that initiates recombinational repair from potentially lethal double strand breaks in DNA which may result from ionizing radiation, replication errors, endonucleases, oxidative damage, and a host of other factors. The RecBCD enzyme is both a helicase that unwinds, or separates the strands of DNA, and a nuclease that makes single-stranded nicks in DNA.

The nucleoid is an irregularly shaped region within the prokaryotic cell that contains all or most of the genetic material. The chromosome of a prokaryote is circular, and its length is very large compared to the cell dimensions needing it to be compacted in order to fit. In contrast to the nucleus of a eukaryotic cell, it is not surrounded by a nuclear membrane. Instead, the nucleoid forms by condensation and functional arrangement with the help of chromosomal architectural proteins and RNA molecules as well as DNA supercoiling. The length of a genome widely varies and a cell may contain multiple copies of it.
A DNA construct is an artificially-designed segment of DNA borne on a vector that can be used to incorporate genetic material into a target tissue or cell. A DNA construct contains a DNA insert, called a transgene, delivered via a transformation vector which allows the insert sequence to be replicated and/or expressed in the target cell. This gene can be cloned from a naturally occurring gene, or synthetically constructed. The vector can be delivered using physical, chemical or viral methods. Typically, the vectors used in DNA constructs contain an origin of replication, a multiple cloning site, and a selectable marker. Certain vectors can carry additional regulatory elements based on the expression system involved.

RecA is a 38 kilodalton protein essential for the repair and maintenance of DNA. A RecA structural and functional homolog has been found in every species in which one has been seriously sought and serves as an archetype for this class of homologous DNA repair proteins. The homologous protein is called RAD51 in eukaryotes and RadA in archaea.
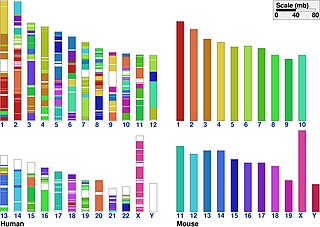
In classical genetics, synteny describes the physical co-localization of genetic loci on the same chromosome within an individual or species. Today, however, biologists usually refer to synteny as the conservation of blocks of order within two sets of chromosomes that are being compared with each other. This concept can also be referred to as shared synteny.

Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids. It is widely used by cells to accurately repair harmful breaks that occur on both strands of DNA, known as double-strand breaks (DSB), in a process called homologous recombinational repair (HRR). Homologous recombination also produces new combinations of DNA sequences during meiosis, the process by which eukaryotes make gamete cells, like sperm and egg cells in animals. These new combinations of DNA represent genetic variation in offspring, which in turn enables populations to adapt during the course of evolution. Homologous recombination is also used in horizontal gene transfer to exchange genetic material between different strains and species of bacteria and viruses.
Synthetic genomics is a nascent field of synthetic biology that uses aspects of genetic modification on pre-existing life forms, or artificial gene synthesis to create new DNA or entire lifeforms.

Artificial gene synthesis, or gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.

In molecular biology, mutagenesis is an important laboratory technique whereby DNA mutations are deliberately engineered to produce libraries of mutant genes, proteins, strains of bacteria, or other genetically modified organisms. The various constituents of a gene, as well as its regulatory elements and its gene products, may be mutated so that the functioning of a genetic locus, process, or product can be examined in detail. The mutation may produce mutant proteins with interesting properties or enhanced or novel functions that may be of commercial use. Mutant strains may also be produced that have practical application or allow the molecular basis of a particular cell function to be investigated.
Synthetic genome is a synthetically-built genome whose formation involves either genetic modification on pre-existing life forms or artificial gene synthesis to create new DNA or entire lifeforms. The field that studies synthetic genomes is called Synthetic Genomics.
SCAR-less genome editing Scarless Cas9 Assisted Recombineering (no-SCAR) is an editing method that is able to manipulate the Escherichia coli genome. The system relies on recombineering whereby DNA sequences are combined and manipulated through homologous recombination. No-SCAR is able to manipulate the E. coli genome without the use of the chromosomal markers detailed in previous recombineering methods. Instead, in this method, the λ-Red recombination system facilitates donor DNA integration while Cas9 cleaves double-stranded DNA to counter-select against wild-type cells. Although λ-Red and Cas9 genome editing are widely used technologies, the no-SCAR method is novel in combining the two functions; this technique is able to establish point mutations, gene deletions, and short sequence insertions in several genomic loci with increased efficiency and time sensitivity.
Bacterial recombination is a type of genetic recombination in bacteria characterized by DNA transfer from one organism called donor to another organism as recipient. This process occurs in three main ways:

Illegitimate recombination, or nonhomologous recombination, is the process by which two unrelated double stranded segments of DNA are joined. This insertion of genetic material which is not meant to be adjacent tends to lead to genes being broken causing the protein which they encode to not be properly expressed. One of the primary pathways by which this will occur is the repair mechanism known as non-homologous end joining (NHEJ).