Related Research Articles
Atomic, molecular, and optical physics (AMO) is the study of matter–matter and light–matter interactions, at the scale of one or a few atoms and energy scales around several electron volts. The three areas are closely interrelated. AMO theory includes classical, semi-classical and quantum treatments. Typically, the theory and applications of emission, absorption, scattering of electromagnetic radiation (light) from excited atoms and molecules, analysis of spectroscopy, generation of lasers and masers, and the optical properties of matter in general, fall into these categories.

A quantum mechanical system or particle that is bound—that is, confined spatially—can only take on certain discrete values of energy, called energy levels. This contrasts with classical particles, which can have any amount of energy. The term is commonly used for the energy levels of the electrons in atoms, ions, or molecules, which are bound by the electric field of the nucleus, but can also refer to energy levels of nuclei or vibrational or rotational energy levels in molecules. The energy spectrum of a system with such discrete energy levels is said to be quantized.

Gerhard Heinrich Friedrich Otto Julius Herzberg, was a German-Canadian pioneering physicist and physical chemist, who won the Nobel Prize for Chemistry in 1971, "for his contributions to the knowledge of electronic structure and geometry of molecules, particularly free radicals". Herzberg's main work concerned atomic and molecular spectroscopy. He is well known for using these techniques that determine the structures of diatomic and polyatomic molecules, including free radicals which are difficult to investigate in any other way, and for the chemical analysis of astronomical objects. Herzberg served as Chancellor of Carleton University in Ottawa, Canada from 1973 to 1980.
Rotational–vibrational spectroscopy is a branch of molecular spectroscopy that is concerned with infrared and Raman spectra of molecules in the gas phase. Transitions involving changes in both vibrational and rotational states can be abbreviated as rovibrational transitions. When such transitions emit or absorb photons, the frequency is proportional to the difference in energy levels and can be detected by certain kinds of spectroscopy. Since changes in rotational energy levels are typically much smaller than changes in vibrational energy levels, changes in rotational state are said to give fine structure to the vibrational spectrum. For a given vibrational transition, the same theoretical treatment as for pure rotational spectroscopy gives the rotational quantum numbers, energy levels, and selection rules. In linear and spherical top molecules, rotational lines are found as simple progressions at both higher and lower frequencies relative to the pure vibration frequency. In symmetric top molecules the transitions are classified as parallel when the dipole moment change is parallel to the principal axis of rotation, and perpendicular when the change is perpendicular to that axis. The ro-vibrational spectrum of the asymmetric rotor water is important because of the presence of water vapor in the atmosphere.

Rotational spectroscopy is concerned with the measurement of the energies of transitions between quantized rotational states of molecules in the gas phase. The rotational spectrum of polar molecules can be measured in absorption or emission by microwave spectroscopy or by far infrared spectroscopy. The rotational spectra of non-polar molecules cannot be observed by those methods, but can be observed and measured by Raman spectroscopy. Rotational spectroscopy is sometimes referred to as pure rotational spectroscopy to distinguish it from rotational-vibrational spectroscopy where changes in rotational energy occur together with changes in vibrational energy, and also from ro-vibronic spectroscopy where rotational, vibrational and electronic energy changes occur simultaneously.
Molecular physics is the study of the physical properties of molecules and molecular dynamics. The field overlaps significantly with physical chemistry, chemical physics, and quantum chemistry. It is often considered as a sub-field of atomic, molecular, and optical physics. Research groups studying molecular physics are typically designated as one of these other fields. Molecular physics addresses phenomena due to both molecular structure and individual atomic processes within molecules. Like atomic physics, it relies on a combination of classical and quantum mechanics to describe interactions between electromagnetic radiation and matter. Experiments in the field often rely heavily on techniques borrowed from atomic physics, such as spectroscopy and scattering.
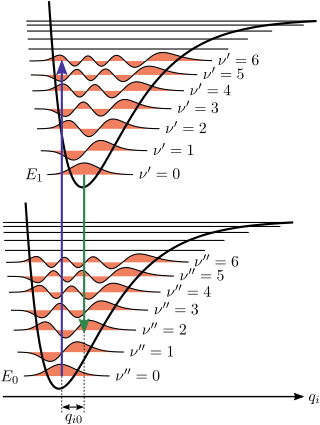
The Franck–Condon principle is a rule in spectroscopy and quantum chemistry that explains the intensity of vibronic transitions. The principle states that during an electronic transition, a change from one vibrational energy level to another will be more likely to happen if the two vibrational wave functions overlap more significantly.
Vibronic coupling in a molecule involves the interaction between electronic and nuclear vibrational motion. The term "vibronic" originates from the combination of the terms "vibrational" and "electronic", denoting the idea that in a molecule, vibrational and electronic interactions are interrelated and influence each other. The magnitude of vibronic coupling reflects the degree of such interrelation.
The Jahn–Teller effect is an important mechanism of spontaneous symmetry breaking in molecular and solid-state systems which has far-reaching consequences in different fields, and is responsible for a variety of phenomena in spectroscopy, stereochemistry, crystal chemistry, molecular and solid-state physics, and materials science. The effect is named for Hermann Arthur Jahn and Edward Teller, who first reported studies about it in 1937.
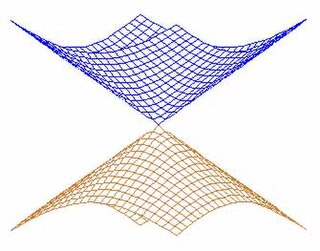
In quantum chemistry, a conical intersection of two or more potential energy surfaces is the set of molecular geometry points where the potential energy surfaces are degenerate (intersect) and the non-adiabatic couplings between these states are non-vanishing. In the vicinity of conical intersections, the Born–Oppenheimer approximation breaks down and the coupling between electronic and nuclear motion becomes important, allowing non-adiabatic processes to take place. The location and characterization of conical intersections are therefore essential to the understanding of a wide range of important phenomena governed by non-adiabatic events, such as photoisomerization, photosynthesis, vision and the photostability of DNA. The conical intersection involving the ground electronic state potential energy surface of the C6H3F3+ molecular ion is discussed in connection with the Jahn–Teller effect in Section 13.4.2 on pages 380-388 of the textbook by Bunker and Jensen.
Rovibronic coupling, also known as rotation/vibration-electron coupling, denotes the simultaneous interactions between rotational, vibrational, and electronic degrees of freedom in a molecule. When a rovibronic transition occurs, the rotational, vibrational, and electronic states change simultaneously, unlike in rovibrational coupling. The coupling can be observed using spectroscopy, and is most easily seen in the Renner–Teller effect in which a linear polyatomic molecule is in a degenerate electronic state and bending vibrations will cause a large rovibronic coupling.
In atomic, molecular, and optical physics and quantum chemistry, the molecular Hamiltonian is the Hamiltonian operator representing the energy of the electrons and nuclei in a molecule. This operator and the associated Schrödinger equation play a central role in computational chemistry and physics for computing properties of molecules and aggregates of molecules, such as thermal conductivity, specific heat, electrical conductivity, optical, and magnetic properties, and reactivity.
A molecular vibration is a periodic motion of the atoms of a molecule relative to each other, such that the center of mass of the molecule remains unchanged. The typical vibrational frequencies range from less than 1013 Hz to approximately 1014 Hz, corresponding to wavenumbers of approximately 300 to 3000 cm−1 and wavelengths of approximately 30 to 3 µm.
An electronic effect influences the structure, reactivity, or properties of a molecule but is neither a traditional bond nor a steric effect. In organic chemistry, the term stereoelectronic effect is also used to emphasize the relation between the electronic structure and the geometry (stereochemistry) of a molecule.
In chemistry, the rotational partition function relates the rotational degrees of freedom to the rotational part of the energy.
Vibronic spectroscopy is a branch of molecular spectroscopy concerned with vibronic transitions: the simultaneous changes in electronic and vibrational energy levels of a molecule due to the absorption or emission of a photon of the appropriate energy. In the gas phase, vibronic transitions are accompanied by changes in rotational energy also.
The pseudo Jahn–Teller effect (PJTE), occasionally also known as second-order JTE, is a direct extension of the Jahn–Teller effect (JTE) where spontaneous symmetry breaking in polyatomic systems occurs even when the relevant electronic states are not degenerate. The PJTE can occur under the influence of sufficiently low-lying electronic excited states of appropriate symmetry. "The pseudo Jahn–Teller effect is the only source of instability and distortions of high-symmetry configurations of polyatomic systems in nondegenerate states, and it contributes significantly to the instability in degenerate states".

Philip R. Bunker is a British-Canadian scientist and author, known for his work in theoretical chemistry and molecular spectroscopy.

Jim Watson, FRS, who published under the name J.K.G. Watson, was a molecular spectroscopist most well known for the development of the widely used molecular Hamiltonians named after him. These Hamiltonians are used to describe the quantum dynamics of molecules.
Second-order Jahn-Teller distortion is a singular, general, and powerful approach rigorously based in first-principle vibronic coupling interactions. It enables prediction and explication of molecular geometries that are not necessarily satisfactorily or even correctly explained by semi-empirical theories such as Walsh diagrams, atomic state hybridization, valence shell electron pair repulsion (VSEPR), softness-hardness-based models, aromaticity and antiaromaticity, hyperconjugation, etc.
References
- 1 2 3 Renner, R. (1934). "Zur Theorie der Wechselwirkung zwischen Elektronen- und Kernbewegung bei dreiatomigen, stabförmigen Molekülen". Zeitschrift für Physik. 92 (3–4): 172–193. Bibcode:1934ZPhy...92..172R. doi:10.1007/BF01350054. S2CID 121493398.
- ↑ Herzberg, G.; Teller, E. (1933). "Schwingungsstruktur der Elektronenübergänge bei mehratomigen Molekülen". Zeitschrift für Physikalische Chemie. 21B: 410–446. doi:10.1515/zpch-1933-2136. S2CID 99159187.
- ↑ Molecular Spectra and Molecular Structure Vol. III, G. Herzberg, Reprint Edition, Krieger, Malabar (1991)
- ↑ Dressler, K.; Ramsay, D. A. (1959). "The electronic absorption spectra of NH2 and ND2". Philosophical Transactions of the Royal Society of London. Series A, Mathematical and Physical Sciences. 251 (1002): 553–602. Bibcode:1959RSPTA.251..553D. doi:10.1098/rsta.1959.0011. S2CID 83464357.
- ↑ Molecular Symmetry and Spectroscopy, 2nd ed. Philip R. Bunker and Per Jensen, NRC Research Press, Ottawa (1998) ISBN 9780660196282