Related Research Articles

Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data, in particular when the data sets are large and complex. As an interdisciplinary field of science, bioinformatics combines biology, chemistry, physics, computer science, information engineering, mathematics and statistics to analyze and interpret the biological data. Bioinformatics has been used for in silico analyses of biological queries using computational and statistical techniques.

Genomics is an interdisciplinary field of biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
The Wellcome Sanger Institute, previously known as The Sanger Centre and Wellcome Trust Sanger Institute, is a non-profit British genomics and genetics research institute, primarily funded by the Wellcome Trust.
The Human Genome Project (HGP) was an international scientific research project with the goal of determining the base pairs that make up human DNA, and of identifying, mapping and sequencing all of the genes of the human genome from both a physical and a functional standpoint. It remains the world's largest collaborative biological project. Planning started after the idea was picked up in 1984 by the US government, the project formally launched in 1990, and was declared essentially complete on April 14, 2003, but included only about 85% of the genome. Level "complete genome" was achieved in May 2021, with a remaining only 0.3% bases covered by potential issues. The final gapless assembly was finished in January 2022.

Elongation factor G 1, mitochondrial is a protein that in humans is encoded by the GFM1 gene. It is an EF-G homolog.

DEAD (Asp-Glu-Ala-Asp) box polypeptide 27, also known as DDX27, is a human gene.
The Centre for Applied Genomics is a genome centre in the Research Institute of The Hospital for Sick Children, and is affiliated with the University of Toronto. TCAG also operates as a Science and Technology Innovation Centre of Genome Canada, with an emphasis on next-generation sequencing (NGS) and bioinformatics support. Research at TCAG focuses on the genetic and genomic basis of human variability, health and disease, including research on the genetics of autism spectrum disorder and structural variation of the human genome. The centre is located in the Peter Gilgan Centre for Research and Learning in downtown Toronto, Canada.

RNA-Seq is a sequencing technique which uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment, analyzing the continuously changing cellular transcriptome.
GENCODE is a scientific project in genome research and part of the ENCODE scale-up project.

Richard Michael Durbin is a British computational biologist and Al-Kindi Professor of Genetics at the University of Cambridge. He also serves as an associate faculty member at the Wellcome Sanger Institute where he was previously a senior group leader.
DECIPHER is a web-based resource and database of genomic variation data from analysis of patient DNA. It documents submicroscopic chromosome abnormalities and pathogenic sequence variants, from over 25000 patients and maps them to the human genome using Ensembl or UCSC Genome Browser. In addition it catalogues the clinical characteristics from each patient and maintains a database of microdeletion/duplication syndromes, together with links to relevant scientific reports and support groups.
GeneNetwork is a combined database and open-source bioinformatics data analysis software resource for systems genetics. This resource is used to study gene regulatory networks that link DNA sequence differences to corresponding differences in gene and protein expression and to variation in traits such as health and disease risk. Data sets in GeneNetwork are typically made up of large collections of genotypes and phenotypes from groups of individuals, including humans, strains of mice and rats, and organisms as diverse as Drosophila melanogaster, Arabidopsis thaliana, and barley. The inclusion of genotypes makes it practical to carry out web-based gene mapping to discover those regions of genomes that contribute to differences among individuals in mRNA, protein, and metabolite levels, as well as differences in cell function, anatomy, physiology, and behavior.
DNA annotation or genome annotation is the process of identifying the locations of genes and all of the coding regions in a genome and determining what those genes do. An annotation is a note added by way of explanation or commentary. Once a genome is sequenced, it needs to be annotated to make sense of it. Genes in a eukaryotic genome can be annotated using various annotation tools such as FINDER. A modern annotation pipeline can support a user-friendly web interface and software containerization such as MOSGA.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.

Jumping libraries or junction-fragment libraries are collections of genomic DNA fragments generated by chromosome jumping. These libraries allow the analysis of large areas of the genome and overcome distance limitations in common cloning techniques. A jumping library clone is composed of two stretches of DNA that are usually located many kilobases away from each other. The stretch of DNA located between these two "ends" is deleted by a series of biochemical manipulations carried out at the start of this cloning technique.
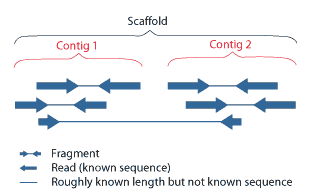
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
The Centre for Genomic Regulation is a biomedical and genomics research centre based on Barcelona. Most of its facilities and laboratories are located in the Barcelona Biomedical Research Park, in front of Somorrostro beach.
References
- ↑ Mott, R; Grigoriev, A; Maier, E; Hoheisel, J; Lehrach, H (1993). "Algorithms and software tools for ordering clone libraries: application to the mapping of the genome of Schizosaccharomyces pombe". Nucleic Acids Res. 21 (8): 1965–74. doi:10.1093/nar/21.8.1965. PMC 309439 . PMID 8493107.
- ↑ Dear et al Genome Res. 1998, 8:260-7
- ↑ Mott, R (1997). "EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA". Comput Appl Biosci. 13 (4): 477–8. doi: 10.1093/bioinformatics/13.4.477 . PMID 9283765.
- ↑ "The Wellcome Centre for Human Genetics — Wellcome Centre for Human Genetics".
- ↑ "Archived copy". Archived from the original on 12 June 2013. Retrieved 2 May 2010.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Mott et al Proc Natl Acad Sci U S A. 2000;97:12649-54
- ↑ Yalcin at al Nat Genet. 2004, 36:1197-202
- ↑ Kover et al PLoS Genet. 2009 Jul;5(7):e1000551