Genetic drift, also known as random genetic drift, allelic drift or the Wright effect, is the change in the frequency of an existing gene variant (allele) in a population due to random chance.
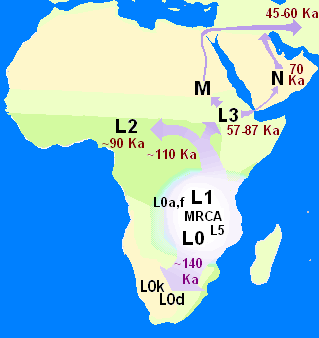
In human genetics, the Mitochondrial Eve is the matrilineal most recent common ancestor (MRCA) of all living humans. In other words, she is defined as the most recent woman from whom all living humans descend in an unbroken line purely through their mothers and through the mothers of those mothers, back until all lines converge on one woman.

The modern synthesis was the early 20th-century synthesis of Charles Darwin's theory of evolution and Gregor Mendel's ideas on heredity into a joint mathematical framework. Julian Huxley coined the term in his 1942 book, Evolution: The Modern Synthesis. The synthesis combined the ideas of natural selection, Mendelian genetics, and population genetics. It also related the broad-scale macroevolution seen by palaeontologists to the small-scale microevolution of local populations.

The neutral theory of molecular evolution holds that most evolutionary changes occur at the molecular level, and most of the variation within and between species are due to random genetic drift of mutant alleles that are selectively neutral. The theory applies only for evolution at the molecular level, and is compatible with phenotypic evolution being shaped by natural selection as postulated by Charles Darwin.
Population genetics is a subfield of genetics that deals with genetic differences within and among populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.

Pseudogenes are nonfunctional segments of DNA that resemble functional genes. Most arise as superfluous copies of functional genes, either directly by gene duplication or indirectly by reverse transcription of an mRNA transcript. Pseudogenes are usually identified when genome sequence analysis finds gene-like sequences that lack regulatory sequences needed for transcription or translation, or whose coding sequences are obviously defective due to frameshifts or premature stop codons. Pseudogenes are a type of junk DNA.
The molecular clock is a figurative term for a technique that uses the mutation rate of biomolecules to deduce the time in prehistory when two or more life forms diverged. The biomolecular data used for such calculations are usually nucleotide sequences for DNA, RNA, or amino acid sequences for proteins.
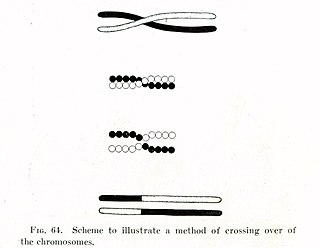
In evolutionary genetics, Muller's ratchet is a process which, in the absence of recombination, results in an accumulation of irreversible deleterious mutations. This happens because in the absence of recombination, and assuming reverse mutations are rare, offspring bear at least as much mutational load as their parents. Muller proposed this mechanism as one reason why sexual reproduction may be favored over asexual reproduction, as sexual organisms benefit from recombination and consequent elimination of deleterious mutations. The negative effect of accumulating irreversible deleterious mutations may not be prevalent in organisms which, while they reproduce asexually, also undergo other forms of recombination. This effect has also been observed in those regions of the genomes of sexual organisms that do not undergo recombination.

Motoo Kimura was a Japanese biologist best known for introducing the neutral theory of molecular evolution in 1968. He became one of the most influential theoretical population geneticists. He is remembered in genetics for his innovative use of diffusion equations to calculate the probability of fixation of beneficial, deleterious, or neutral alleles. Combining theoretical population genetics with molecular evolution data, he also developed the neutral theory of molecular evolution in which genetic drift is the main force changing allele frequencies. James F. Crow, himself a renowned population geneticist, considered Kimura to be one of the two greatest evolutionary geneticists, along with Gustave Malécot, after the great trio of the modern synthesis, Ronald Fisher, J. B. S. Haldane, and Sewall Wright.

Sewall Green Wright FRS (For) Honorary FRSE was an American geneticist known for his influential work on evolutionary theory and also for his work on path analysis. He was a founder of population genetics alongside Ronald Fisher and J. B. S. Haldane, which was a major step in the development of the modern synthesis combining genetics with evolution. He discovered the inbreeding coefficient and methods of computing it in pedigree animals. He extended this work to populations, computing the amount of inbreeding between members of populations as a result of random genetic drift, and along with Fisher he pioneered methods for computing the distribution of gene frequencies among populations as a result of the interaction of natural selection, mutation, migration and genetic drift. Wright also made major contributions to mammalian and biochemical genetics.
Coalescent theory is a model of how alleles sampled from a population may have originated from a common ancestor. In the simplest case, coalescent theory assumes no recombination, no natural selection, and no gene flow or population structure, meaning that each variant is equally likely to have been passed from one generation to the next. The model looks backward in time, merging alleles into a single ancestral copy according to a random process in coalescence events. Under this model, the expected time between successive coalescence events increases almost exponentially back in time. Variance in the model comes from both the random passing of alleles from one generation to the next, and the random occurrence of mutations in these alleles.
In genetics, the Ka/Ks ratio, also known as ω or dN/dS ratio, is used to estimate the balance between neutral mutations, purifying selection and beneficial mutations acting on a set of homologous protein-coding genes. It is calculated as the ratio of the number of nonsynonymous substitutions per non-synonymous site (Ka), in a given period of time, to the number of synonymous substitutions per synonymous site (Ks), in the same period. The latter are assumed to be neutral, so that the ratio indicates the net balance between deleterious and beneficial mutations. Values of Ka/Ks significantly above 1 are unlikely to occur without at least some of the mutations being advantageous. If beneficial mutations are assumed to make little contribution, then Ka/Ks estimates the degree of evolutionary constraint.
Neutral mutations are changes in DNA sequence that are neither beneficial nor detrimental to the ability of an organism to survive and reproduce. In population genetics, mutations in which natural selection does not affect the spread of the mutation in a species are termed neutral mutations. Neutral mutations that are inheritable and not linked to any genes under selection will be lost or will replace all other alleles of the gene. That loss or fixation of the gene proceeds based on random sampling known as genetic drift. A neutral mutation that is in linkage disequilibrium with other alleles that are under selection may proceed to loss or fixation via genetic hitchhiking and/or background selection.

Masatoshi Nei was a Japanese-born American evolutionary biologist.
Background selection describes the loss of genetic diversity at a locus due to negative selection against deleterious alleles with which it is in linkage disequilibrium. The name emphasizes the fact that the genetic background, or genomic environment, of a mutation has a significant impact on whether it will be preserved versus lost from a population. Background selection contradicts the assumption of the neutral theory of molecular evolution that the fixation or loss of a neutral allele can be described by one-locus models of genetic drift, independently from other loci. As well as reducing neutral nucleotide diversity, background selection reduces the fixation probability of beneficial mutations, and increases the fixation probability of deleterious mutations.
A genetic lineage includes all descendants of a given genetic sequence, typically following a new mutation. It is not the same as an allele because it excludes cases where different mutations give rise to the same allele, and includes descendants that differ from the ancestor by one or more mutations. The genetic sequence can be of different sizes, e.g. a single gene or a haplotype containing multiple adjacent genes along a chromosome. Given recombination, each gene can have a separate genetic lineages, even as the population shares a single organismal lineage. In asexual microbes or somatic cells, cell lineages exactly match genetic lineages, and can be traced.
Listed here are notable ethnic groups and native populations from the Oceania and East Indonesia by human Y-chromosome DNA haplogroups based on relevant studies.
The McDonald–Kreitman test is a statistical test often used by evolutionary and population biologists to detect and measure the amount of adaptive evolution within a species by determining whether adaptive evolution has occurred, and the proportion of substitutions that resulted from positive selection. To do this, the McDonald–Kreitman test compares the amount of variation within a species (polymorphism) to the divergence between species (substitutions) at two types of sites, neutral and nonneutral. A substitution refers to a nucleotide that is fixed within one species, but a different nucleotide is fixed within a second species at the same base pair of homologous DNA sequences. A site is nonneutral if it is either advantageous or deleterious. The two types of sites can be either synonymous or nonsynonymous within a protein-coding region. In a protein-coding sequence of DNA, a site is synonymous if a point mutation at that site would not change the amino acid, also known as a silent mutation. Because the mutation did not result in a change in the amino acid that was originally coded for by the protein-coding sequence, the phenotype, or the observable trait, of the organism is generally unchanged by the silent mutation. A site in a protein-coding sequence of DNA is nonsynonymous if a point mutation at that site results in a change in the amino acid, resulting in a change in the organism's phenotype. Typically, silent mutations in protein-coding regions are used as the "control" in the McDonald–Kreitman test.
Viral phylodynamics is the study of how epidemiological, immunological, and evolutionary processes act and potentially interact to shape viral phylogenies. Since the term was coined in 2004, research on viral phylodynamics has focused on transmission dynamics in an effort to shed light on how these dynamics impact viral genetic variation. Transmission dynamics can be considered at the level of cells within an infected host, individual hosts within a population, or entire populations of hosts.
Brandon Stuart Gaut is an American evolutionary biologist and geneticist who works as a Distinguished Professor of Ecology and Evolutionary Biology at the University of California, Irvine.