How SOSUI works
First of all, SOSUI looks for α helices that are relatively easy to predict, taking into account the known helical potentials of the given amino acid sequence(AAS). The much more difficult task is to differentiate between the α helices in soluble proteins and the ones in transmembrane proteins, the α helix being a very common secondary structure pattern in proteins. SOSUI uses 4 characteristics of the AAS in its prediction:
- "hydropathy index" (Kyte und Doolittle 1982)
- weighted presence of amphiphilic amino acids (AA) and their localization: "amphiphilicity index"
- the AA's charge
- the length of the AAS
An important improvement compared to Kyte und Doolittle's "hydropathy index", which relies entirely on one characteristic, is the introduction of the so-called "amphiphilicity index". It is calculated by giving every AA with an amphiphilic residue a certain value which is derived from the AA's molecular structure. To meet SOSUI's criteria for amphiphilicity, the polar, hydrophilic residue may not be linked directly to the beta-carbon; there must be at least one apolar carbon interposed (therefore only lysine, arginine, histidine, glutamic acid, glutamine, tryptophan and tyrosine are relevant). SOSUI then looks for accumulations of amphiphilic AAs at the ends of α helices, which seems to be typical for transmembrane α helices (it makes the transmembrane position the energetically best one for these α helices by placing amphiphilic AAs at the lipid-water boundary and is thus co-responsible for the protein's correct localization). The AA's charge is also taken into consideration; the length is important because biological lipid membranes have a certain thickness determining the length of membrane-spanning proteins. According to a study published by SOSUI's developers it successfully differentiated 99% of a chosen group of proteins with known structure . However, another study that had several prediction tools perform on the AAS's of 122 known proteins claimed that SOSUI was correct about the number of α helices in only about 60% of the cases . But even if the number of transmembrane domains is not always exact, the differentiation between soluble and transmembrane proteins often works, as it is only necessary to find out if a protein has such a domain at all. Of course, membrane proteins which don't have transmembrane α helices (e.g. porins) or which are fixed with a covalent bond cannot be found by SOSUI.

The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group hydrogen bonds to the backbone C=O group of the amino acid located four residues earlier along the protein sequence.
Transmembrane domain usually denotes a transmembrane segment of single alpha helix of a transmembrane protein. More broadly, a transmembrane domain is any membrane-spanning protein domain.
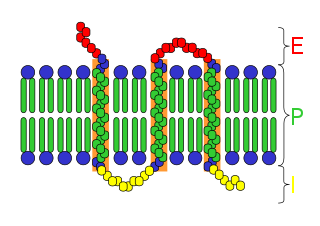
An integral membrane protein (IMP) is a type of membrane protein that is permanently attached to the biological membrane. All transmembrane proteins are IMPs, but not all IMPs are transmembrane proteins. IMPs comprise a significant fraction of the proteins encoded in an organism's genome. Proteins that cross the membrane are surrounded by annular lipids, which are defined as lipids that are in direct contact with a membrane protein. Such proteins can only be separated from the membranes by using detergents, nonpolar solvents, or sometimes denaturing agents.

Membrane proteins are common proteins that are part of, or interact with, biological membranes. Membrane proteins fall into several broad categories depending on their location. Integral membrane proteins are a permanent part of a cell membrane and can either penetrate the membrane (transmembrane) or associate with one or the other side of a membrane. Peripheral membrane proteins are transiently associated with the cell membrane.

A transmembrane protein (TP) is a type of integral membrane protein that spans the entirety of the cell membrane. Many transmembrane proteins function as gateways to permit the transport of specific substances across the membrane. They frequently undergo significant conformational changes to move a substance through the membrane. They are usually highly hydrophobic and aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents.

Peripheral membrane proteins are membrane proteins that adhere only temporarily to the biological membrane with which they are associated. These proteins attach to integral membrane proteins, or penetrate the peripheral regions of the lipid bilayer. The regulatory protein subunits of many ion channels and transmembrane receptors, for example, may be defined as peripheral membrane proteins. In contrast to integral membrane proteins, peripheral membrane proteins tend to collect in the water-soluble component, or fraction, of all the proteins extracted during a protein purification procedure. Proteins with GPI anchors are an exception to this rule and can have purification properties similar to those of integral membrane proteins.

Topology of a transmembrane protein refers to locations of N- and C-termini of membrane-spanning polypeptide chain with respect to the inner or outer sides of the biological membrane occupied by the protein.
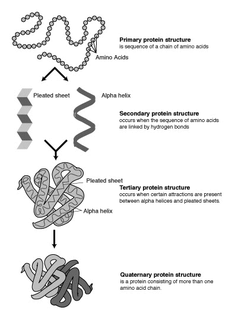
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
Orientations of Proteins in Membranes (OPM) database provides spatial positions of membrane protein structures with respect to the lipid bilayer. Positions of the proteins are calculated using an implicit solvation model of the lipid bilayer. The results of calculations were verified against experimental studies of spatial arrangement of transmembrane and peripheral proteins in membranes.
Hydrophobicity scales are values that define the relative hydrophobicity or hydrophilicity of amino acid residues. The more positive the value, the more hydrophobic are the amino acids located in that region of the protein. These scales are commonly used to predict the transmembrane alpha-helices of membrane proteins. When consecutively measuring amino acids of a protein, changes in value indicate attraction of specific protein regions towards the hydrophobic region inside lipid bilayer.

The Bcl-2 family consists of a number of evolutionarily-conserved proteins that share Bcl-2 homology (BH) domains. The Bcl-2 family is most notable for their regulation of apoptosis, a form of programmed cell death, at the mitochondrion. The Bcl-2 family proteins consists of members that either promote or inhibit apoptosis, and control apoptosis by governing mitochondrial outer membrane permeabilization (MOMP), which is a key step in the intrinsic pathway of apoptosis. A total of 25 genes in the Bcl-2 family were identified by 2008.

WALP peptides are a class of synthesized, membrane-spanning α-helices composed of tryptophan (W), alanine (A), and leucine (L) amino acids. They are designed to study properties of proteins in lipid membranes such as orientation, extent of insertion, and hydrophobic mismatch.
The Hopp–Woods hydrophilicity scale of amino acids is a method of ranking the amino acids in a protein according to their water solubility in order to search for surface locations on proteins, and especially those locations that tend to form strong interactions with other macromolecules such as proteins, DNA, and RNA.

Transmembrane protein 131-like, alternatively named uncharacterized protein KIAA0922, is an integral transmembrane protein encoded by the human gene KIAA0922 that is significantly conserved in eukaryotes, at least through protists. Although the function of this gene is not yet fully elucidated, initial microarray evidence suggests that it may be involved in immune responses. Furthermore, its paralog, prolyl endopeptidase (PREP) whose function is known, provides clues as to the function of TMEM131L.
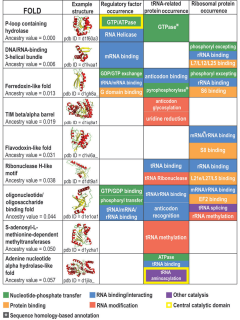
Protein fold classes are broad categories of protein tertiary structure topology. They describe groups of proteins that share similar amino acid and secondary structure proportions. Each class contains multiple, independent protein superfamilies.

Collagen α-1 (XXIII) chain is a protein encoded by COL23A1 gene, which is located on chromosome 5q35 in humans, and on chromosome 11B1+2 in mice. The location of this gene was discovered by genomic sequence analysis.
Transmembrane protein 251, also known as C14orf109 or UPF0694, is a protein that in humans is encoded by the TMEM251 gene. One notable feature of this protein is the presence of proline residues on one of its predicted transmembrane domains., which is a determinant of the intramitochondrial sorting of inner membrane proteins.
Membranome database provides structural and functional information about more than 6000 single-pass (bitopic) transmembrane proteins from Homo sapiens, Arabidopsis thaliana, Dictyostelium discoideum, Saccharomyces cerevisiae, Escherichia coli and Methanocaldococcus jannaschii. Bitopic membrane proteins consist of a single transmembrane alpha-helix connecting water-soluble domains of the protein situated at the opposite sides of a biological membrane. These proteins are frequently involved in the signal transduction and communication between cells in multicellular organisms.

C16orf90 or chromosome 16 open reading frame 90 produces uncharacterized protein C16orf90 in homo sapiens. C16orf90's protein has four predicted alpha-helix domains and is mildly expressed in the testes and lowly expressed throughout the body. While the function of C16orf90 is not yet well understood by the scientific community, it has suspected involvement in the biological stress response and apoptosis based on expression data from microarrays and post-translational modification data.
The QTY Code is a design method to transform membrane proteins that are intrinsically insoluble in water into variants with water solubility, while retaining their structure and function.