
Pharmacology is the science of drugs and medications, including a substance's origin, composition, pharmacokinetics, pharmacodynamics, therapeutic use, and toxicology. More specifically, it is the study of the interactions that occur between a living organism and chemicals that affect normal or abnormal biochemical function. If substances have medicinal properties, they are considered pharmaceuticals.
Pharmacovigilance, also known as drug safety, is the pharmaceutical science relating to the "collection, detection, assessment, monitoring, and prevention" of adverse effects with pharmaceutical products. The etymological roots for the word "pharmacovigilance" are: pharmakon and vigilare. As such, pharmacovigilance heavily focuses on adverse drug reactions (ADR), which are defined as any response to a drug which is noxious and unintended, including lack of efficacy. Medication errors such as overdose, and misuse and abuse of a drug as well as drug exposure during pregnancy and breastfeeding, are also of interest, even without an adverse event, because they may result in an adverse drug reaction.

The Common Technical Document (CTD) is a set of specifications for an application dossier for the registration of medicine, designed for use across Europe, Japan, the United States, and beyond.

The European Medicines Agency (EMA) is an agency of the European Union (EU) in charge of the evaluation and supervision of pharmaceutical products. Prior to 2004, it was known as the European Agency for the Evaluation of Medicinal Products or European Medicines Evaluation Agency (EMEA).
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is an initiative that brings together regulatory authorities and pharmaceutical industry to discuss scientific and technical aspects of pharmaceutical product development and registration. The mission of the ICH is to promote public health by achieving greater harmonisation through the development of technical guidelines and requirements for pharmaceutical product registration.
A serious adverse event (SAE) in human drug trials is defined as any untoward medical occurrence that at any dose
- Results in death
- Is life-threatening
- Requires inpatient hospitalization or causes prolongation of existing hospitalization
- Results in persistent or significant disability/incapacity
- May have caused a congenital anomaly/birth defect
- Requires intervention to prevent permanent impairment or damage
Good clinical practice (GCP) is an international quality standard, which governments can then transpose into regulations for clinical trials involving human subjects. GCP follows the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), and enforces tight guidelines on ethical aspects of clinical research.

The Council for International Organizations of Medical Sciences (CIOMS) is an international non-governmental organization of 40 international, national, and associate member groups representing the biomedical science community. It was jointly established by the World Health Organization (WHO) and United Nations Educational, Scientific and Cultural Organization (UNESCO) in 1949 as a successor to the International Medical Congress that organized 17 conferences from 1867 until the 1913 outbreak of World War I.
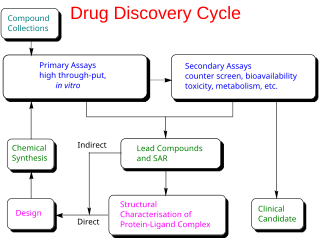
Drug development is the process of bringing a new pharmaceutical drug to the market once a lead compound has been identified through the process of drug discovery. It includes preclinical research on microorganisms and animals, filing for regulatory status, such as via the United States Food and Drug Administration for an investigational new drug to initiate clinical trials on humans, and may include the step of obtaining regulatory approval with a new drug application to market the drug. The entire process—from concept through preclinical testing in the laboratory to clinical trial development, including Phase I–III trials—to approved vaccine or drug typically takes more than a decade.
A standard operating procedure (SOP) is a set of step-by-step instructions compiled by an organization to help workers carry out routine operations. SOPs aim to achieve efficiency, quality output, and uniformity of performance, while reducing miscommunication and failure to comply with industry regulations.
The International Union of Basic and Clinical Pharmacology (IUPHAR) is a voluntary, non-profit association representing the interests of scientists in pharmacology-related fields to facilitate Better Medicines through Global Education and Research around the world.

Uppsala Monitoring Centre (UMC), located in Uppsala, Sweden, is the field name for the World Health Organization Collaborating Centre for International Drug Monitoring. UMC works by collecting, assessing and communicating information from member countries' national pharmacovigilance centres in regard to the benefits, harm, effectiveness and risks of drugs.
In drug development and medical device development the Investigator's Brochure (IB) is a comprehensive document summarizing the body of information about an investigational product obtained during a drug trial. The IB is a document of critical importance throughout the drug development process and is updated with new information as it becomes available. The purpose of the IB is to compile data relevant to studies of the IP in human subjects gathered during preclinical and other clinical trials.
A clinical investigator involved in a clinical trial is responsible for ensuring that an investigation is conducted according to the signed investigator statement, the investigational plan, and applicable regulations; for protecting the rights, safety, and welfare of subjects under the investigator's care; and for the control of drugs under investigation. The Clinical Investigator must also meet requirements set forth by the FDA, EMA or other regulatory body. The qualifications must be outlined in a current resume and readily available for auditors.

The European Directorate for the Quality of Medicines & HealthCare (EDQM) is a Directorate and partial agreement of the Council of Europe that traces its origins and statutes to the Convention on the Elaboration of a European Pharmacopoeia.
Safety pharmacology is a branch of pharmacology specialising in detecting and investigating potential undesirable pharmacodynamic effects of new chemical entities (NCEs) on physiological functions in relation to exposure in the therapeutic range and above.

Syed Ziaur Rahman is a permanent member of 'Board of Trustees' and Chair of the Advisory Council, International Association of Medical Colleges (IAOMC). He also serves as Chairman, Department of Pharmacology, Jawaharlal Nehru Medical College, Aligarh, Elected Secretary of IAOMC and Society of Pharmacovigilance, India (SoPI).
Donald Robert James Singer was a British clinical pharmacologist who was the president of the Fellowship of Postgraduate Medicine.
In medicine, a clinical study report (CSR) on a clinical trial is a document, typically very long, providing much detail about the methods and results of a trial. A CSR is a scientific document addressing efficacy and safety, not a sales or marketing tool; its content is similar to that of a peer-reviewed academic paper. Results of trials are usually reported in a briefer academic journal paper, but methodological flaws are often glossed over in the briefer paper.
Guidances for statistics in regulatory affairs refers to specific documents or guidelines that provide instructions, recommendations, and standards pertaining to the application of statistical methodologies and practices within the regulatory framework of industries such as pharmaceuticals and medical devices. These guidances serve as a reference for statisticians, researchers, and professionals involved in designing, conducting, analyzing, and reporting studies and trials in compliance with regulatory requirements. These documents embody the prevailing perspectives of regulatory agencies on specific subjects. It is worth noting that in the United States, the term "Guidances" is used, while in Europe, the term "Guidelines" is employed.