
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.

Molecular evolution is the process of change in the sequence composition of cellular molecules such as DNA, RNA, and proteins across generations. The field of molecular evolution uses principles of evolutionary biology and population genetics to explain patterns in these changes. Major topics in molecular evolution concern the rates and impacts of single nucleotide changes, neutral evolution vs. natural selection, origins of new genes, the genetic nature of complex traits, the genetic basis of speciation, evolution of development, and ways that evolutionary forces influence genomic and phenotypic changes.
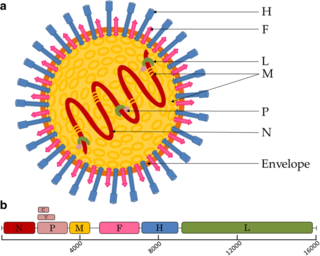
Paramyxoviridae is a family of negative-strand RNA viruses in the order Mononegavirales. Vertebrates serve as natural hosts. Diseases associated with this family include measles, mumps, and respiratory tract infections. The family has four subfamilies, 17 genera, and 78 species, three genera of which are unassigned to a subfamily.
The coding region of a gene, also known as the coding sequence(CDS), is the portion of a gene's DNA or RNA that codes for protein. Studying the length, composition, regulation, splicing, structures, and functions of coding regions compared to non-coding regions over different species and time periods can provide a significant amount of important information regarding gene organization and evolution of prokaryotes and eukaryotes. This can further assist in mapping the human genome and developing gene therapy.
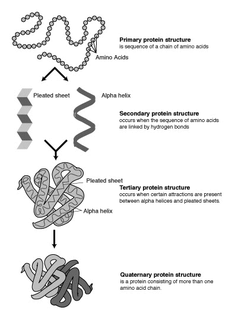
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.

In genetics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.

Peripherin is a type III intermediate filament protein expressed mainly in neurons of the peripheral nervous system. It is also found in neurons of the central nervous system that have projections toward peripheral structures, such as spinal motor neurons. Its size, structure, and sequence/location of protein motifs is similar to other type III intermediate filament proteins such as desmin, vimentin and glial fibrillary acidic protein. Like these proteins, peripherin can self-assemble to form homopolymeric filamentous networks, but it can also heteropolymerize with neurofilaments in several neuronal types. This protein in humans is encoded by the PRPH gene. Peripherin is thought to play a role in neurite elongation during development and axonal regeneration after injury, but its exact function is unknown. It is also associated with some of the major neuropathologies that characterize amyotropic lateral sclerosis (ALS), but despite extensive research into how neurofilaments and peripherin contribute to ALS, their role in this disease is still unidentified.
In evolutionary biology, conserved sequences are identical or similar sequences in nucleic acids or proteins across species, or within a genome, or between donor and receptor taxa. Conservation indicates that a sequence has been maintained by natural selection.

In biology, the word gene can have several different meanings. The Mendelian gene is a basic unit of heredity and the molecular gene is a sequence of nucleotides in DNA that is transcribed to produce a functional RNA. There are two types of molecular genes: protein-coding genes and noncoding genes.
InterPro is a database of protein families, protein domains and functional sites in which identifiable features found in known proteins can be applied to new protein sequences in order to functionally characterise them.

H5N1 genetic structure is the molecular structure of the H5N1 virus's RNA.
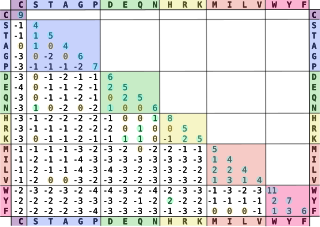
In bioinformatics, the BLOSUM matrix is a substitution matrix used for sequence alignment of proteins. BLOSUM matrices are used to score alignments between evolutionarily divergent protein sequences. They are based on local alignments. BLOSUM matrices were first introduced in a paper by Steven Henikoff and Jorja Henikoff. They scanned the BLOCKS database for very conserved regions of protein families and then counted the relative frequencies of amino acids and their substitution probabilities. Then, they calculated a log-odds score for each of the 210 possible substitution pairs of the 20 standard amino acids. All BLOSUM matrices are based on observed alignments; they are not extrapolated from comparisons of closely related proteins like the PAM Matrices.
Antigenic variation or antigenic alteration refers to the mechanism by which an infectious agent such as a protozoan, bacterium or virus alters the proteins or carbohydrates on its surface and thus avoids a host immune response, making it one of the mechanisms of antigenic escape. It is related to phase variation. Antigenic variation not only enables the pathogen to avoid the immune response in its current host, but also allows re-infection of previously infected hosts. Immunity to re-infection is based on recognition of the antigens carried by the pathogen, which are "remembered" by the acquired immune response. If the pathogen's dominant antigen can be altered, the pathogen can then evade the host's acquired immune system. Antigenic variation can occur by altering a variety of surface molecules including proteins and carbohydrates. Antigenic variation can result from gene conversion, site-specific DNA inversions, hypermutation, or recombination of sequence cassettes. The result is that even a clonal population of pathogens expresses a heterogeneous phenotype. Many of the proteins known to show antigenic or phase variation are related to virulence.

HIKESHI is a protein important in lung and multicellular organismal development that, in humans, is encoded by the HIKESHI gene. HIKESHI is found on chromosome 11 in humans and chromosome 7 in mice. Similar sequences (orthologs) are found in most animal and fungal species. The mouse homolog, lethal gene on chromosome 7 Rinchik 6 protein is encoded by the l7Rn6 gene.
The Virus Pathogen Database and Analysis Resource (ViPR) is an integrative and comprehensive publicly available database and analysis resource to search, analyze, visualize, save and share data for viral pathogens in the U.S. National Institute of Allergy and Infectious Diseases (NIAID) Category A-C Priority Pathogen lists for biodefense research, and other viral pathogens causing emerging/reemerging infectious diseases. ViPR is one of the five Bioinformatics Resource Centers (BRC) funded by NIAID, a component of the National Institutes of Health (NIH), which is an agency of the United States Department of Health and Human Services.
The Influenza Research Database (IRD) is an integrative and comprehensive publicly available database and analysis resource to search, analyze, visualize, save and share data for influenza virus research. IRD is one of the five Bioinformatics Resource Centers (BRC) funded by the National Institute of Allergy and Infectious Diseases (NIAID), a component of the National Institutes of Health (NIH), which is an agency of the United States Department of Health and Human Services.

Transmembrane protein 131-like, alternatively named uncharacterized protein KIAA0922, is an integral transmembrane protein encoded by the human gene KIAA0922 that is significantly conserved in eukaryotes, at least through protists. Although the function of this gene is not yet fully elucidated, initial microarray evidence suggests that it may be involved in immune responses. Furthermore, its paralog, prolyl endopeptidase (PREP) whose function is known, provides clues as to the function of TMEM131L.
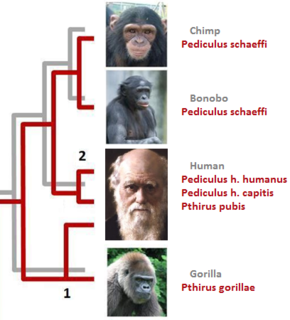
In parasitology and epidemiology, a host switch is an evolutionary change of the host specificity of a parasite or pathogen. For example, the human immunodeficiency virus used to infect and circulate in non-human primates in West-central Africa, but switched to humans in the early 20th century.