
In biology, a mutation is an alteration in the nucleic acid sequence of the genome of an organism, virus, or extrachromosomal DNA. Viral genomes contain either DNA or RNA. Mutations result from errors during DNA or viral replication, mitosis, or meiosis or other types of damage to DNA, which then may undergo error-prone repair, cause an error during other forms of repair, or cause an error during replication. Mutations may also result from insertion or deletion of segments of DNA due to mobile genetic elements.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesizes the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.
Protein engineering is the process of developing useful or valuable proteins. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.
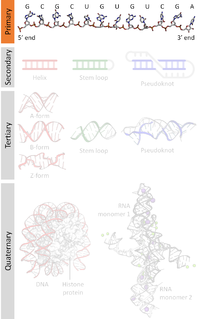
A nucleic acid sequence is a succession of bases signified by a series of a set of five different letters that indicate the order of nucleotides forming alleles within a DNA or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.
Molecular genetics is a sub-field of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens. The field of study is based on the merging of several sub-fields in biology: classical Mendelian inheritance, cellular biology, molecular biology, biochemistry, and biotechnology. Researchers search for mutations in a gene or induce mutations in a gene to link a gene sequence to a specific phenotype. Molecular genetics is a powerful methodology for linking mutations to genetic conditions that may aid the search for treatments/cures for various genetics diseases.
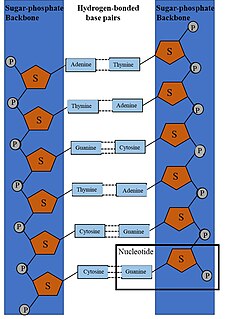
DNA synthesis is the natural or artificial creation of deoxyribonucleic acid (DNA) molecules. DNA is a macromolecule made up of nucleotide units, which are linked by covalent bonds and hydrogen bonds, in a repeating structure. DNA synthesis occurs when these nucleotide units are joined to form DNA; this can occur artificially or naturally. Nucleotide units are made up of a nitrogenous base, pentose sugar (deoxyribose) and phosphate group. Each unit is joined when a covalent bond forms between its phosphate group and the pentose sugar of the next nucleotide, forming a sugar-phosphate backbone. DNA is a complementary, double stranded structure as specific base pairing occurs naturally when hydrogen bonds form between the nucleotide bases.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.

A point mutation is a genetic mutation where a single nucleotide base is changed, inserted or deleted from a DNA or RNA sequence of an organism's genome. Point mutations have a variety of effects on the downstream protein product—consequences that are moderately predictable based upon the specifics of the mutation. These consequences can range from no effect to deleterious effects, with regard to protein production, composition, and function.

In molecular biology, a library is a collection of DNA fragments that is stored and propagated in a population of micro-organisms through the process of molecular cloning. There are different types of DNA libraries, including cDNA libraries, genomic libraries and randomized mutant libraries. DNA library technology is a mainstay of current molecular biology, genetic engineering, and protein engineering, and the applications of these libraries depend on the source of the original DNA fragments. There are differences in the cloning vectors and techniques used in library preparation, but in general each DNA fragment is uniquely inserted into a cloning vector and the pool of recombinant DNA molecules is then transferred into a population of bacteria or yeast such that each organism contains on average one construct. As the population of organisms is grown in culture, the DNA molecules contained within them are copied and propagated.
In genetics, a missense mutation is a point mutation in which a single nucleotide change results in a codon that codes for a different amino acid. It is a type of nonsynonymous substitution.

In genetics, an insertion is the addition of one or more nucleotide base pairs into a DNA sequence. This can often happen in microsatellite regions due to the DNA polymerase slipping. Insertions can be anywhere in size from one base pair incorrectly inserted into a DNA sequence to a section of one chromosome inserted into another. The mechanism of the smallest single base insertion mutations is believed to be through base-pair separation between the template and primer strands followed by non-neighbor base stacking, which can occur locally within the DNA polymerase active site. On a chromosome level, an insertion refers to the insertion of a larger sequence into a chromosome. This can happen due to unequal crossover during meiosis.

Directed evolution (DE) is a method used in protein engineering that mimics the process of natural selection to steer proteins or nucleic acids toward a user-defined goal. It consists of subjecting a gene to iterative rounds of mutagenesis, selection and amplification. It can be performed in vivo, or in vitro. Directed evolution is used both for protein engineering as an alternative to rationally designing modified proteins, as well as for experimental evolution studies of fundamental evolutionary principles in a controlled, laboratory environment.
In genetics, the Ka/Ks ratio, also known as ω or dN/dS ratio, is used to estimate the balance between neutral mutations, purifying selection and beneficial mutations acting on a set of homologous protein-coding genes. It is calculated as the ratio of the number of nonsynonymous substitutions per non-synonymous site (Ka), in a given period of time, to the number of synonymous substitutions per synonymous site (Ks), in the same period. The latter are assumed to be neutral, so that the ratio indicates the net balance between deleterious and beneficial mutations. Values of Ka/Ks significantly above 1 are unlikely to occur without at least some of the mutations being advantageous. If beneficial mutations are assumed to make little contribution, then Ka/Ks estimates the degree of evolutionary constraint.
Missense mRNA is a messenger RNA bearing one or more mutated codons that yield polypeptides with an amino acid sequence different from the wild-type or naturally occurring polypeptide. Missense mRNA molecules are created when template DNA strands or the mRNA strands themselves undergo a missense mutation in which a protein coding sequence is mutated and an altered amino acid sequence is coded for.
Site saturation mutagenesis (SSM), or simply site saturation, is a random mutagenesis technique used in protein engineering, in which a single codon or set of codons is substituted with all possible amino acids at the position. There are many variants of the site saturation technique, from paired site saturation to scanning site saturation.

In molecular biology, mutagenesis is an important laboratory technique whereby DNA mutations are deliberately engineered to produce libraries of mutant genes, proteins, strains of bacteria, or other genetically modified organisms. The various constituents of a gene, as well as its regulatory elements and its gene products, may be mutated so that the functioning of a genetic locus, process, or product can be examined in detail. The mutation may produce mutant proteins with interesting properties or enhanced or novel functions that may be of commercial use. Mutant strains may also be produced that have practical application or allow the molecular basis of a particular cell function to be investigated.
Degeneracy or redundancy of codons is the redundancy of the genetic code, exhibited as the multiplicity of three-base pair codon combinations that specify an amino acid. The degeneracy of the genetic code is what accounts for the existence of synonymous mutations.

SeSaM-Biotech GmbH is a biotechnology service company founded in 2008 in Bremen and localized in Aachen today.
Ulrich Schwaneberg is a German chemist and protein engineer. He is the Chair of Biotechnology at RWTH Aachen University and member of the scientific board at the Leibniz Institute for Interactive Materials in Aachen. He specializes in directed evolution of proteins for material science applications and on the development of its methodologies. The latter comprise methods for diversity generation, as well as high-throughput screening systems. His work group has elucidated general design principles of enzymes by analyzing libraries that contain the full natural diversity of a hydrolase with single amino acid exchanges and developed strategies to efficiently explore the protein sequence space and discovered protein engineering principles.
Genetic saturation is the result of multiple substitutions at the same site in a sequence, or identical substitutions in different sequences, such that the apparent sequence divergence rate is lower than the actual divergence that has occurred. When comparing two or more genetic sequences consisting of single nucleotides, differences in sequence observed are only differences in the final state of the nucleotide sequence. Single nucleotides that undergoing genetic saturation change multiple times, sometimes back to their original nucleotide or to a nucleotide common to the compared genetic sequence. Without genetic information from intermediate taxa, it is difficult to know how much, or if any saturation has occurred on an observed sequence. Genetic saturation occurs most rapidly on fast-evolving sequences, such as the hypervariable region of mitochondrial DNA, or in short tandem repeats such as on the Y-chromosome.

