Related Research Articles

DNA ligase is a type of enzyme that facilitates the joining of DNA strands together by catalyzing the formation of a phosphodiester bond. It plays a role in repairing single-strand breaks in duplex DNA in living organisms, but some forms may specifically repair double-strand breaks. Single-strand breaks are repaired by DNA ligase using the complementary strand of the double helix as a template, with DNA ligase creating the final phosphodiester bond to fully repair the DNA.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. A synthetic primer may also be referred to as an oligo, short for oligonucleotide. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesizes the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.

DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.

Rolling circle replication (RCR) is a process of unidirectional nucleic acid replication that can rapidly synthesize multiple copies of circular molecules of DNA or RNA, such as plasmids, the genomes of bacteriophages, and the circular RNA genome of viroids. Some eukaryotic viruses also replicate their DNA or RNA via the rolling circle mechanism.
The selector technique is a method to amplify and multiplex genomic DNA.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
An allele-specific oligonucleotide (ASO) is a short piece of synthetic DNA complementary to the sequence of a variable target DNA. It acts as a probe for the presence of the target in a Southern blot assay or, more commonly, in the simpler dot blot assay. It is a common tool used in genetic testing, forensics, and molecular biology research.

SOLiD (Sequencing by Oligonucleotide Ligation and Detection) is a next-generation DNA sequencing technology developed by Life Technologies and has been commercially available since 2006. This next generation technology generates 108 - 109 small sequence reads at one time. It uses 2 base encoding to decode the raw data generated by the sequencing platform into sequence data.
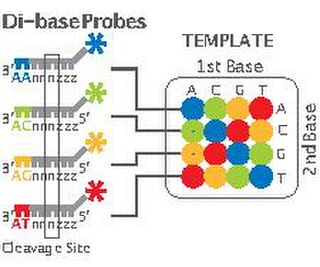
2 Base Encoding, also called SOLiD, is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing, the Solexa system and the SOLiD system. These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.

Oligomer Restriction is a procedure to detect an altered DNA sequence in a genome. A labeled oligonucleotide probe is hybridized to a target DNA, and then treated with a restriction enzyme. If the probe exactly matches the target, the restriction enzyme will cleave the probe, changing its size. If, however, the target DNA does not exactly match the probe, the restriction enzyme will have no effect on the length of the probe. The OR technique, now rarely performed, was closely associated with the development of the popular polymerase chain reaction (PCR) method.
The versatility of polymerase chain reaction (PCR) has led to modifications of the basic protocol being used in a large number of variant techniques designed for various purposes. This article summarizes many of the most common variations currently or formerly used in molecular biology laboratories; familiarity with the fundamental premise by which PCR works and corresponding terms and concepts is necessary for understanding these variant techniques.
The ligase chain reaction (LCR) is a method of DNA amplification. The ligase chain reaction (LCR) is an amplification process that differs from PCR in that it involves a thermostable ligase to join two probes or other molecules together which can then be amplified by standard polymerase chain reaction (PCR) cycling. Each cycle results in a doubling of the target nucleic acid molecule. A key advantage of LCR is greater specificity as compared to PCR. Thus, LCR requires two completely different enzymes to operate properly: ligase, to join probe molecules together, and a thermostable polymerase to amplify those molecules involved in successful ligation. The probes involved in the ligation are designed such that the 5′ end of one probe is directly adjacent to the 3′ end of the other probe, thereby providing the requisite 3′-OH and 5′-PO4 group substrates for the ligase.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.
DNA ends refer to the properties of the ends of linear DNA molecules, which in molecular biology are described as "sticky" or "blunt" based on the shape of the complementary strands at the terminus. In sticky ends, one strand is longer than the other, such that the longer strand has bases which are left unpaired. In blunt ends, both strands are of equal length – i.e. they end at the same base position, leaving no unpaired bases on either strand.

Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.
Magnetic sequencing is a single-molecule sequencing method in development. A DNA hairpin, containing the sequence of interest, is bound between a magnetic bead and a glass surface. A magnetic field is applied to stretch the hairpin open into single strands, and the hairpin refolds after decreasing of the magnetic field. The hairpin length can be determined by direct imaging of the diffraction rings of the magnetic beads using a simple microscope. The DNA sequences are determined by measuring the changes in the hairpin length following successful hybridization of complementary nucleotides.
A hybridization assay comprises any form of quantifiable hybridization i.e. the quantitative annealing of two complementary strands of nucleic acids, known as nucleic acid hybridization.
This glossary of genetics is a list of definitions of terms and concepts commonly used in the study of genetics and related disciplines in biology, including molecular biology, cell biology, and evolutionary biology. It is intended as introductory material for novices; for more specific and technical detail, see the article corresponding to each term. For related terms, see Glossary of evolutionary biology.
References
- ↑ S. C. Macevicz, US Patent 5750341, filed 1995
- ↑ Whiteley (1988). "Detection of specific sequences in nucleic acids". US Patent 4,883,750.
- ↑ J. Shendure, G.J. Porreca, N.B. Reppas, X. Lin, J.Pe McCutcheon, A.M. Rosenbaum, M.D. Wang, K. Zhang, R.D. Mitra and G.M. Church (2005). "Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome". Science. 309 (5741): 1728–1732. Bibcode:2005Sci...309.1728S. doi: 10.1126/science.1117389 . PMID 16081699. S2CID 11405973.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Yu-Feng Huang, Sheng-Chung Chen, Yih-Shien Chiang, Tzu-Han Chen & Kuo-Ping Chiu (2012). "Palindromic sequence impedes sequencing-by-ligation mechanism". BMC Systems Biology . 6 (Suppl 2): S10. doi: 10.1186/1752-0509-6-S2-S10 . PMC 3521181 . PMID 23281822.
{{cite journal}}: CS1 maint: multiple names: authors list (link)