Achondroplasia is a genetic disorder with an autosomal dominant pattern of inheritance whose primary feature is dwarfism. In those with the condition, the arms and legs are short, while the torso is typically of normal length. Those affected have an average adult height of 131 centimetres for males and 123 centimetres (4 ft) for females. Other features can include an enlarged head and prominent forehead. Complications can include sleep apnea or recurrent ear infections. Achondroplasia includes the extremely rare short-limb skeletal dysplasia with severe combined immunodeficiency.

Dwarfism is a condition wherein an organism is exceptionally small, and mostly occurs in the animal kingdom. In humans, it is sometimes defined as an adult height of less than 147 centimetres, regardless of sex; the average adult height among people with dwarfism is 120 centimetres (4 ft). Disproportionate dwarfism is characterized by either short limbs or a short torso. In cases of proportionate dwarfism, both the limbs and torso are unusually small. Intelligence is usually normal, and most have a nearly normal life expectancy. People with dwarfism can usually bear children, though there are additional risks to the mother and child depending upon the underlying condition.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Since the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects.

Thanatophoric dysplasia is a severe skeletal disorder characterized by a disproportionately small ribcage, extremely short limbs and folds of extra skin on the arms and legs.

Hypochondroplasia (HCH) is a developmental disorder caused by an autosomal dominant genetic defect in the fibroblast growth factor receptor 3 gene (FGFR3) that results in a disproportionately short stature, micromelia and a head that appears large in comparison with the underdeveloped portions of the body. It is classified as short-limbed dwarfism.
Crouzonodermoskeletal syndrome is a disorder characterized by the premature joining of certain bones of the skull (craniosynostosis) during development and a skin condition called acanthosis nigricans.
Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.
Osteochondrodysplasia is a general term for a disorder of the development (dysplasia) of bone ("osteo") and cartilage ("chondro"). Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. Osteochondrodysplasias can result in marked functional limitation and even mortality.

Pseudoachondroplasia is an inherited disorder of bone growth. It is a genetic autosomal dominant disorder. It is generally not discovered until 2–3 years of age, since growth is normal at first. Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities.
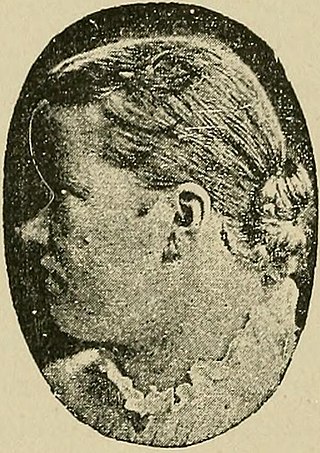
Muenke syndrome, also known as FGFR3-related craniosynostosis, is a human specific condition characterized by the premature closure of certain bones of the skull during development, which affects the shape of the head and face. First described by Maximilian Muenke, the syndrome occurs in about 1 in 30,000 newborns. This condition accounts for an estimated 8 percent of all cases of craniosynostosis.

Fibroblast growth factor receptor 3 is a protein that in humans is encoded by the FGFR3 gene. FGFR3 has also been designated as CD333. The gene, which is located on chromosome 4, location p16.3, is expressed in tissues such as the cartilage, brain, intestine, and kidneys.

Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.
3-M syndrome or 3M3 is a rare hereditary disorder characterized by severe growth retardation, facial dysmorphia, and skeletal abnormalities. The name 3-M is derived from the initials of the three researchers who first identified it: Miller, McKusick, and Malvaux and report their findings in the medical literature in 1972. Mutations in any one of the following three genes: CUL7, OBSL1, and CCDC8 are responsible for the occurrence of this disorder. It is inherited through an autosomal recessive pattern and considered very rare, so far less than 100 cases worldwide have been identified. Diagnosis is based on the presence of clinical features. Genetic testing can confirm the diagnosis and identify the specific gene involved. Treatment is aimed at addressing the growth and skeletal problems and may include surgical bone lengthening, adaptive aids, and physical therapy. An endocrinologist may assist with growth hormone replacement and appropriate evaluations during puberty.
Zoltan Vajo is a Hungarian/American scientist, best known for his contributions to the Human Genome Project, including cloning the COQ7 gene, characterizing the human CLK-1 timing protein cDNA and its potential effect on aging, and research on the molecular and genetic background of skeletal dysplasias and fibroblast growth factor receptor 3 disorders, including Achondroplasia, SADDAN, Thanatophoric dysplasia, Muenke coronal craniosynostosis and Crouzon syndrome as well as more recently on genetically engineered insulin analog molecules, including their structure, metabolic effects and cellular processing and the role of recombinant DNA technology in the treatment of diabetes.
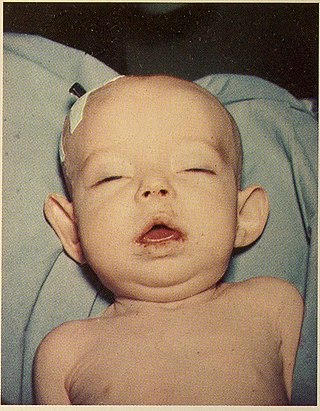
Donohue syndrome is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors. As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia, hypoglycemia, hyperinsulemia, and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
Beare–Stevenson cutis gyrata syndrome is a rare genetic disorder characterized by craniosynostosis and a specific skin abnormality, called cutis gyrata, characterized by a furrowed and wrinkled appearance ; thick, dark, velvety areas of skin are sometimes found on the hands and feet and in the groin.

Baller–Gerold syndrome (BGS) is a rare genetic syndrome that involves premature fusion of the skull bones and malformations of facial, forearm and hand bones. The symptoms of Baller–Gerold syndrome overlap with features of a few other genetics disorders: Rothmund–Thomson syndrome and RAPADILINO syndrome. The prevalence of BGS is unknown, as there have only been a few reported cases, but it is estimated to be less than 1 in a million. The name of the syndrome comes from the researchers Baller and Gerold who discovered the first three cases.
Opitz G/BBB syndrome, also known as Opitz syndrome, G syndrome or BBB syndrome, is a rare genetic disorder that will affect physical structures along the midline of the body. The letters G and BBB represent the last names of the families that were first diagnosed with the disorder, while Opitz is the last name of the doctor that first described the signs and symptoms of the disease. There are two different forms of Optiz G/BBB syndrome: x-linked (recessive) syndrome and dominant autosomal syndrome. However, both result in common physical deformities, although their pattern of inheritance may differ. Several other names for the disease(s) are no longer used. These include hypospadias-dysphagia syndrome, Opitz-Frias syndrome, telecanthus with associated abnormalities, and hypertelorism-hypospadias syndrome.
Clair A. Francomano is an American medical geneticist and academic specializing in Ehlers–Danlos syndromes. She is Professor of Medical and Molecular Genetics at Indiana University.
