An allele, or allelomorph, is a variant of the sequence of nucleotides at a particular location, or locus, on a DNA molecule.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.

In genetics, dominance is the phenomenon of one variant (allele) of a gene on a chromosome masking or overriding the effect of a different variant of the same gene on the other copy of the chromosome. The first variant is termed dominant and the second is called recessive. This state of having two different variants of the same gene on each chromosome is originally caused by a mutation in one of the genes, either new or inherited. The terms autosomal dominant or autosomal recessive are used to describe gene variants on non-sex chromosomes (autosomes) and their associated traits, while those on sex chromosomes (allosomes) are termed X-linked dominant, X-linked recessive or Y-linked; these have an inheritance and presentation pattern that depends on the sex of both the parent and the child. Since there is only one copy of the Y chromosome, Y-linked traits cannot be dominant or recessive. Additionally, there are other forms of dominance, such as incomplete dominance, in which a gene variant has a partial effect compared to when it is present on both chromosomes, and co-dominance, in which different variants on each chromosome both show their associated traits.
Genetic drift, also known as random genetic drift, allelic drift or the Wright effect, is the change in the frequency of an existing gene variant (allele) in a population due to random chance.
Population genetics is a subfield of genetics that deals with genetic differences within and among populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.
Allele frequency, or gene frequency, is the relative frequency of an allele at a particular locus in a population, expressed as a fraction or percentage. Specifically, it is the fraction of all chromosomes in the population that carry that allele over the total population or sample size. Microevolution is the change in allele frequencies that occurs over time within a population.
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.

In biology, polymorphism is the occurrence of two or more clearly different morphs or forms, also referred to as alternative phenotypes, in the population of a species. To be classified as such, morphs must occupy the same habitat at the same time and belong to a panmictic population.

A haplotype is a group of alleles in an organism that are inherited together from a single parent.

Motoo Kimura was a Japanese biologist best known for introducing the neutral theory of molecular evolution in 1968. He became one of the most influential theoretical population geneticists. He is remembered in genetics for his innovative use of diffusion equations to calculate the probability of fixation of beneficial, deleterious, or neutral alleles. Combining theoretical population genetics with molecular evolution data, he also developed the neutral theory of molecular evolution in which genetic drift is the main force changing allele frequencies. James F. Crow, himself a renowned population geneticist, considered Kimura to be one of the two greatest evolutionary geneticists, along with Gustave Malécot, after the great trio of the modern synthesis, Ronald Fisher, J. B. S. Haldane, and Sewall Wright.
A null allele is a nonfunctional allele caused by a genetic mutation. Such mutations can cause a complete lack of production of the associated gene product or a product that does not function properly; in either case, the allele may be considered nonfunctional. A null allele cannot be distinguished from deletion of the entire locus solely from phenotypic observation.

Genetic distance is a measure of the genetic divergence between species or between populations within a species, whether the distance measures time from common ancestor or degree of differentiation. Populations with many similar alleles have small genetic distances. This indicates that they are closely related and have a recent common ancestor.
In population genetics, fixation is the change in a gene pool from a situation where there exists at least two variants of a particular gene (allele) in a given population to a situation where only one of the alleles remains. That is, the allele becomes fixed. In the absence of mutation or heterozygote advantage, any allele must eventually be lost completely from the population or fixed. Whether a gene will ultimately be lost or fixed is dependent on selection coefficients and chance fluctuations in allelic proportions. Fixation can refer to a gene in general or particular nucleotide position in the DNA chain (locus).
Marker assisted selection or marker aided selection (MAS) is an indirect selection process where a trait of interest is selected based on a marker linked to a trait of interest, rather than on the trait itself. This process has been extensively researched and proposed for plant- and animal- breeding.
The infinite alleles model is a mathematical model for calculating genetic mutations. The Japanese geneticist Motoo Kimura and American geneticist James F. Crow (1964) introduced the infinite alleles model, an attempt to determine for a finite diploid population what proportion of loci would be homozygous. This was, in part, motivated by assertions by other geneticists that more than 50 percent of Drosophila loci were heterozygous, a claim they initially doubted. In order to answer this question they assumed first, that there were a large enough number of alleles so that any mutation would lead to a different allele ; and second, that the mutations would result in a number of different outcomes from neutral to deleterious.
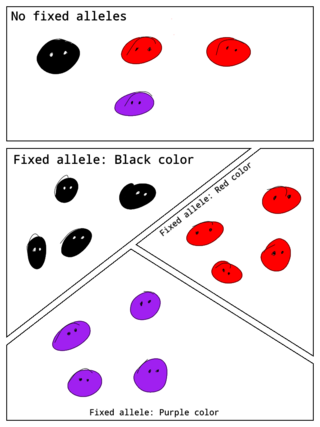
In population genetics, a fixed allele is an allele that is the only variant that exists for that gene in a population. A fixed allele is homozygous for all members of the population. The process by which alleles become fixed is called fixation.

Zygosity is the degree to which both copies of a chromosome or gene have the same genetic sequence. In other words, it is the degree of similarity of the alleles in an organism.

A gene is said to be polymorphic if more than one allele occupies that gene's locus within a population. In addition to having more than one allele at a specific locus, each allele must also occur in the population at a rate of at least 1% to generally be considered polymorphic.
In population genetics, the allele frequency spectrum, sometimes called the site frequency spectrum, is the distribution of the allele frequencies of a given set of loci in a population or sample. Because an allele frequency spectrum is often a summary of or compared to sequenced samples of the whole population, it is a histogram with size depending on the number of sequenced individual chromosomes. Each entry in the frequency spectrum records the total number of loci with the corresponding derived allele frequency. Loci contributing to the frequency spectrum are assumed to be independently changing in frequency. Furthermore, loci are assumed to be biallelic, although extensions for multiallelic frequency spectra exist.
The Infinite sites model (ISM) is a mathematical model of molecular evolution first proposed by Motoo Kimura in 1969. Like other mutation models, the ISM provides a basis for understanding how mutation develops new alleles in DNA sequences. Using allele frequencies, it allows for the calculation of heterozygosity, or genetic diversity, in a finite population and for the estimation of genetic distances between populations of interest.