Related Research Articles
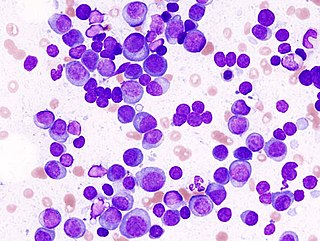
Multiple myeloma (MM), also known as plasma cell myeloma and simply myeloma, is a cancer of plasma cells, a type of white blood cell that normally produces antibodies. Often, no symptoms are noticed initially. As it progresses, bone pain, anemia, renal insufficiency, and infections may occur. Complications may include hypercalcemia and amyloidosis.
Hematopoietic stem-cell transplantation (HSCT) is the transplantation of multipotent hematopoietic stem cells, usually derived from bone marrow, peripheral blood, or umbilical cord blood, in order to replicate inside a patient and produce additional normal blood cells. HSCT may be autologous, syngeneic, or allogeneic.

Hypereosinophilic syndrome is a disease characterized by a persistently elevated eosinophil count in the blood for at least six months without any recognizable cause, with involvement of either the heart, nervous system, or bone marrow.

POEMS syndrome is a rare paraneoplastic syndrome caused by a clone of aberrant plasma cells. The name POEMS is an acronym for some of the disease's major signs and symptoms, as is PEP.
Leukapheresis is a laboratory procedure in which white blood cells are separated from a sample of blood. It is a specific type of apheresis, the more general term for separating out one particular constituent of blood and returning the remainder to the circulation.

Monoclonal gammopathy of undetermined significance (MGUS) is a plasma cell dyscrasia in which plasma cells or other types of antibody-producing cells secrete a myeloma protein, i.e. an abnormal antibody, into the blood; this abnormal protein is usually found during standard laboratory blood or urine tests. MGUS resembles multiple myeloma and similar diseases, but the levels of antibodies are lower, the number of plasma cells in the bone marrow is lower, and it rarely has symptoms or major problems. However, since MGUS can lead to multiple myeloma, which develops at the rate of about 1.5% a year, or other symptomatic conditions, yearly monitoring is recommended.

Hepatorenal syndrome (HRS) is a life-threatening medical condition that consists of rapid deterioration in kidney function in individuals with cirrhosis or fulminant liver failure. HRS is usually fatal unless a liver transplant is performed, although various treatments, such as dialysis, can prevent advancement of the condition.
CHOP is the acronym for a chemotherapy regimen used in the treatment of non-Hodgkin lymphoma. CHOP consists of:

Peliosis hepatis is an uncommon vascular condition characterised by multiple, randomly distributed, blood-filled cavities throughout the liver. The size of the cavities usually ranges between a few millimetres and 3 cm in diameter. In the past, it was a mere histological curiosity occasionally found at autopsies, but has been increasingly recognised with wide-ranging conditions from AIDS to the use of anabolic steroids. It also occasionally affects spleen, lymph nodes, lungs, kidneys, adrenal glands, bone marrow, and other parts of gastrointestinal tract.
In hematology, plasma cell dyscrasias are a spectrum of progressively more severe monoclonal gammopathies in which a clone or multiple clones of pre-malignant or malignant plasma cells over-produce and secrete into the blood stream a myeloma protein, i.e. an abnormal monoclonal antibody or portion thereof. The exception to this rule is the disorder termed non-secretory multiple myeloma; this disorder is a form of plasma cell dyscrasia in which no myeloma protein is detected in serum or urine of individuals who have clear evidence of an increase in clonal bone marrow plasma cells and/or evidence of clonal plasma cell-mediated tissue injury. Here, a clone of plasma cells refers to group of plasma cells that are abnormal in that they have an identical genetic identity and therefore are descendants of a single genetically distinct ancestor cell.
Amyloid light-chain (AL) amyloidosis, also known as primary amyloidosis, is the most common form of systemic amyloidosis. The disease is caused when a person's antibody-producing cells do not function properly and produce abnormal protein fibers made of components of antibodies called light chains. These light chains come together to form amyloid deposits which can cause serious damage to different organs. An abnormal light chain in urine is known as Bence Jones protein.

Plasma cell leukemia (PCL) is a plasma cell dyscrasia, i.e. a disease involving the malignant degeneration of a subtype of white blood cells called plasma cells. It is the terminal stage and most aggressive form of these dyscrasias, constituting 2% to 4% of all cases of plasma cell malignancies. PCL may present as primary plasma cell leukemia, i.e. in patients without prior history of a plasma cell dyscrasia or as secondary plasma cell dyscrasia, i.e. in patients previously diagnosed with a history of its predecessor dyscrasia, multiple myeloma. The two forms of PCL appear to be at least partially distinct from each other. In all cases, however, PCL is an extremely serious, life-threatening, and therapeutically challenging disease.
Fanconi syndrome or Fanconi's syndrome is a syndrome of inadequate reabsorption in the proximal renal tubules of the kidney. The syndrome can be caused by various underlying congenital or acquired diseases, by toxicity, or by adverse drug reactions. It results in various small molecules of metabolism being passed into the urine instead of being reabsorbed from the tubular fluid. Fanconi syndrome affects the proximal tubules, namely, the proximal convoluted tubule (PCT), which is the first part of the tubule to process fluid after it is filtered through the glomerulus, and the proximal straight tubule, which leads to the descending limb of loop of Henle.

Daratumumab, sold under the brand name Darzalex among others, is an anti-cancer monoclonal antibody medication. It binds to CD38, which is overexpressed in multiple myeloma cells. Daratumumab was originally developed by Genmab, but it is now being jointly developed by Genmab along with the Johnson & Johnson subsidiary Janssen Biotech, which acquired worldwide commercialization rights to the drug from Genmab.

Light chain deposition disease (LCDD) is a rare blood cell disease which is characterized by deposition of fragments of infection-fighting immunoglobulins, called light chains (LCs), in the body. LCs are normally cleared by the kidneys, but in LCDD, these light chain deposits damage organs and cause disease. The kidneys are almost always affected and this often leads to kidney failure. About half of people with light chain deposition disease also have a plasma cell dyscrasia, a spectrum of diseases that includes multiple myeloma, Waldenström's macroglobulinemia, and the monoclonal gammopathy of undetermined significance premalignant stages of these two diseases. Unlike in AL amyloidosis, in which light chains are laid down in characteristic amyloid deposits, in LCDD, light chains are deposited in non-amyloid granules.
Onconephrology is a specialty in nephrology that deals with the study of kidney diseases in cancer patients. A nephrologist who takes care of patients with cancer and kidney disease is called an onconephrologist. This branch of nephrology encompasses nephrotoxicity associated with existing and novel chemotherapeutics, kidney disease as it pertains to stem cell transplant, paraneoplastic kidney disorders, paraproteinemias, electrolyte disorders associated with cancer, and more as discussed below.
Daratumumab/hyaluronidase, sold under the brand name Darzalex Faspro, is a fixed-dose combination medication for the treatment of adults with newly diagnosed or relapsed/refractory multiple myeloma. It is a combination of daratumumab and hyaluronidase. It is administered via subcutaneous injection.
Idecabtagene vicleucel, sold under the brand name Abecma, is a cell-based gene therapy to treat multiple myeloma.
Monoclonal immunoglobulin deposition disease, or MIDD, is a disease characterised by the deposition of monoclonal immunoglobulins on the basement membrane of the kidney. Monoclonal immunoglobulins are produced by monoclonal plasma cells, which are found in a variety of plasma cell dyscrasias. The deposition of monoclonal immunoglobulins on the basement membrane of the kidney causes renal impairment. As well as the kidney, MIDD may also affect the liver, heart, peripheral nerves, lung and skin.
Monoclonal gammopathy of renal significance (MGRS) are a group of kidney disorders that present with kidney damage due to nephrotoxic monoclonal immunoglobulins secreted by clonal plasma cells or B cells. By definition, people with MGRS do not meet criteria for multiple myeloma or other hematologic malignancies. The term MGRS was introduced in 2012 by the International Kidney and Monoclonal Gammopathy Research Group (IKMG). MGRS is associated with monoclonal gammopathy of undetermined significance (MGUS). People with MGUS have a monoclonal gammopathy but does not meet the criteria for the clonal burden nor the presence of end organ damage seen in hematologic malignancies. In a population based study based on the NHANES III health survey; 6% of patients with MGUS were subsequently classified as having MGRS. The prevalence and incidence of MGRS in the general population or in specific populations is not known but it is more prevalent in those over the age of 50 as there is a monoclonal protein (M-protein) present in 3% of those 50 and years older and 5% of those 70 years and older, placing those 50 and older at increased risk of MGRS.
References
- ↑ Sykes, David B.; Schroyens, Wilfried; O'Connell, Casey (2011). "TEMPI Syndrome – A Novel Multisystem Disease". N Engl J Med . 365 (5): 475–477. doi: 10.1056/NEJMc1106670 . PMID 21812700. S2CID 35990145.
- 1 2 Sykes, David B.; O'Connell, Casey; Schroyens, Wilfried (2020-04-09). "The TEMPI syndrome". Blood. 135 (15): 1199–1203. doi: 10.1182/blood.2019004216 . ISSN 1528-0020. PMID 32108223.
- ↑ Schroyens, Wilfried; O'Connell, Casey; Sykes, David B. (2012). "Complete and Partial Responses of the TEMPI Syndrome to Bortezomib" (PDF). N Engl J Med . 367 (8): 778–780. doi:10.1056/NEJMc1205806. PMID 22913703.
- ↑ Sykes, David B.; Schroyens, W. (2018). "Complete Responses in the TEMPI Syndrome after Treatment with Daratumumab". N Engl J Med . 378 (23): 2240–2242. doi:10.1056/NEJMc1804415. PMID 29874534. S2CID 205064988.
- ↑ Kenderian, S.S..; Rosado, F.G; Sykes, D.B.; Hoyer, J.D.; Lacy, M.Q. (2015). "Long-term complete clinical and hematological responses of the TEMPI syndrome after autologous stem cell transplantation". Leukemia . 29 (12): 2414–2416. doi: 10.1038/leu.2015.298 . PMID 26500143.
- ↑ Bazari, Hasan; Attar, Eyal C.; Dahl, Douglas M.; Uppot, Raul N.; Colvin, Robert B. (2010). "Case Records of the Massachusetts General Hospital. Case 23-2010: A 49-Year-Old Man with Erythrocytosis, Perinephric Fluid Collections, and Renal Failure". N Engl J Med . 363 (5): 463–475. doi:10.1056/NEJMcpc1004086. PMID 20818867.