
Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data. As an interdisciplinary field of science, bioinformatics combines biology, computer science, information engineering, mathematics and statistics to analyze and interpret biological data. Bioinformatics has been used for in silico analyses of biological queries using mathematical and statistical techniques.
Grammar theory to model symbol strings originated from work in computational linguistics aiming to understand the structure of natural languages. Probabilistic context free grammars (PCFGs) have been applied in probabilistic modeling of RNA structures almost 40 years after they were introduced in computational linguistics.

The SIB Swiss Institute of Bioinformatics is an academic not-for-profit foundation which federates bioinformatics activities throughout Switzerland.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional “gene-by-gene” approach.
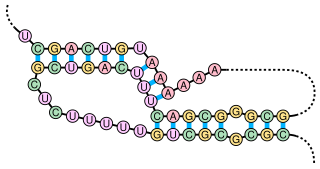
A pseudoknot is a nucleic acid secondary structure containing at least two stem-loop structures in which half of one stem is intercalated between the two halves of another stem. The pseudoknot was first recognized in the turnip yellow mosaic virus in 1982. Pseudoknots fold into knot-shaped three-dimensional conformations but are not true topological knots.
Protein–protein interaction prediction is a field combining bioinformatics and structural biology in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding protein–protein interactions is important for the investigation of intracellular signaling pathways, modelling of protein complex structures and for gaining insights into various biochemical processes.

Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
Nucleic acid structure prediction is a computational method to determine secondary and tertiary nucleic acid structure from its sequence. Secondary structure can be predicted from one or several nucleic acid sequences. Tertiary structure can be predicted from the sequence, or by comparative modeling.
Rfam is a database containing information about non-coding RNA (ncRNA) families and other structured RNA elements. It is an annotated, open access database originally developed at the Wellcome Trust Sanger Institute in collaboration with Janelia Farm, and currently hosted at the European Bioinformatics Institute. Rfam is designed to be similar to the Pfam database for annotating protein families.
HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and Mac OS.
Partition function for Interacting RNAs (piRNA) is a parallel C++ package to compute joint and individual partition functions for two RNA sequences. From the partition functions, piRNA computes equilibrium concentrations of single and double species, ensemble energy, melting temperatures, and base pair probabilities. piRNA is part of TaveRNA RNA software suite.
BioSLAX is a Live CD/Live DVD/Live USB comprising a suite of more than 300 bioinformatics tools and application suites. It has been released by the Bioinformatics Resource Unit of the Life Sciences Institute (LSI), National University of Singapore (NUS) and is bootable from any PC that allows a CD/DVD or USB boot option and runs the compressed Slackware flavour of the Linux Operating System (OS), also known as Slax. Slax was created by Tomáš Matějíček in the Czech Republic using the Linux Live Scripts which he also developed. The BioSLAX derivative was created by Mark De Silva, Lim Kuan Siong and Tan Tin Wee.
GeneCards is a database of human genes that provides genomic, proteomic, transcriptomic, genetic and functional information on all known and predicted human genes. It is being developed and maintained by the Crown Human Genome Center at the Weizmann Institute of Science. This database aims at providing a quick overview of the current available biomedical information about the searched gene, including the human genes, the encoded proteins, and the relevant diseases. The GeneCards database provides access to free Web resources about more than 7000 all known human genes that integrated from >90 data resources, such as HGNC, Ensembl, and NCBI. The core gene list is based on approved gene symbols published by the HUGO Gene Nomenclature Committee (HGNC). The information are carefully gathered and selected from these databases by the powerful and user-friendly engine. If the search does not return any results, this database will give several suggestions to help users accomplish their searching depended on the type of query, and offer direct links to other databases’ search engine. Over time, the GeneCards database has developed a suite of tools that has more specialised capability. Since 1998, the GeneCards database has been widely used by bioinformatics, genomics and medical communities for more than 15 years.

The Nucleic Acid Package (NUPACK) is a growing software suite for the analysis and design of nucleic acid systems. Jobs can be run online on the NUPACK webserver or NUPACK source code can be downloaded and compiled locally for non-commercial academic use. NUPACK algorithms are formulated in terms of nucleic acid secondary structure. In most cases, pseudoknots are excluded from the structural ensemble.
This comparison of DNA melting prediction software includes source code and web based software for predicting DNA melting and structure.
A neutral network is a set of genes all related by point mutations that have equivalent function or fitness. Each node represents a gene sequence and each line represents the mutation connecting two sequences. Neutral networks can be thought of as high, flat plateaus in a fitness landscape. During neutral evolution, genes can randomly move through neutral networks and traverse regions of sequence space which may have consequences for robustness and evolvability.

Gary Stormo is an American geneticist and currently Joseph Erlanger Professor in the Department of Genetics and the Center for Genome Sciences and Systems Biology at Washington University School of Medicine in St Louis. He is considered as one of the pioneers of bioinformatics and genomics. His research combines experimental and computational approaches in order to identify and predict regulatory sequences in DNA and RNA, and their contributions to the regulatory networks that control gene expression.
The ViennaRNA Package is a set of standalone programs and libraries used for prediction and analysis of RNA secondary structures. The source code for the package is distributed freely and compiled binaries are available for Linux, macOS and Windows platforms. The original paper has been cited over 2000 times.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.

