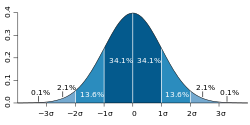
A plot of normal distribution (or bell-shaped curve) where each band has a width of 1 standard deviation. If the threshold is 2 standard deviations above the mean of the latent variable, then about 2.4% of the population would have the trait.
In mathematical or statistical modeling a threshold model is any model where a threshold value, or set of threshold values, is used to distinguish ranges of values where the behaviour predicted by the model varies in some important way. A particularly important instance arises in toxicology, where the model for the effect of a drug may be that there is zero effect for a dose below a critical or threshold value, while an effect of some significance exists above that value.[1] Certain types of regression model may include threshold effects.[1]
Threshold models are often used to model the behavior of groups, ranging from social insects to animal herds to human society.
Classic threshold models were introduced by Sakoda,[2] in his 1949 dissertation and the Journal of Mathematical Sociology (JMS vol 1 #1, 1971).[3] They were subsequently developed by Schelling, Axelrod, and Granovetter to model collective behavior. Schelling used a special case of Sakoda's model to describe the dynamics of segregation motivated by individual interactions in America (JMS vol 1 #2, 1971)[4] by constructing two simulation models. Schelling demonstrated that “there is no simple correspondence of individual incentive to collective results,” and that the dynamics of movement influenced patterns of segregation. In doing so Schelling highlighted the significance of “a general theory of ‘tipping’”.
Mark Granovetter, following Schelling, proposed the threshold model (Granovetter & Soong, 1983, 1986, 1988), which assumes that individuals’ behavior depends on the number of other individuals already engaging in that behavior (both Schelling and Granovetter classify their term of “threshold” as behavioral threshold.). He used the threshold model to explain the riot, residential segregation, and the spiral of silence. In the spirit of Granovetter's threshold model, the “threshold” is “the number or proportion of others who must make one decision before a given actor does so”. It is necessary to emphasize the determinants of threshold. Different individuals have different thresholds. Individuals' thresholds may be influenced by many factors: social economic status, education, age, personality, etc. Further, Granovetter relates “threshold” with utility one gets from participating in collective behavior or not, using the utility function, each individual will calculate his or her cost and benefit from undertaking an action. And situation may change the cost and benefit of the behavior, so threshold is situation-specific. The distribution of the thresholds determines the outcome of the aggregate behavior (for example, public opinion).
A threshold model used in toxicology posits that anything above a certain dose of a toxin is dangerous, and anything below it safe. This model is usually applied to non-carcinogenic health hazards.
The threshold dose-response model is widely viewed as the most dominant model in toxicology.[6]
An alternative type of model in toxicology is the linear no-threshold model (LNT), while hormesis correspond to the existence of opposite effects at low vs. high dose, which usually gives a U- or inverted U-shaped dose response curve.
Liability threshold model
The liability-threshold model is a threshold model of categorical (usually binary) outcomes in which a large number of variables are summed to yield an overall 'liability' score; the observed outcome is determined by whether the latent score is smaller or larger than the threshold. The liability-threshold model is frequently employed in medicine and genetics to model risk factors contributing to disease.
In a genetic context, the variables are all the genes and different environmental conditions, which protect against or increase the risk of a disease, and the threshold z is the biological limit past which disease develops. The threshold can be estimated from population prevalence of the disease (which is usually low). Because the threshold is defined relative to the population & environment, the liability score is generally considered as a N(0, 1) normally distributedrandom variable.
Early genetics models were developed to deal with very rare genetic diseases by treating them as Mendelian diseases caused by 1 or 2 genes: the presence or absence of the gene corresponds to the presence or absence of the disease, and the occurrence of the disease will follow predictable patterns within families. Continuous traits like height or intelligence could be modeled as normal distributions, influenced by a large number of genes, and the heritability and effects of selection easily analyzed. Some diseases, like alcoholism, epilepsy, or schizophrenia, cannot be Mendelian diseases because they are common; do not appear in Mendelian ratios; respond slowly to selection against them; often occur in families with no prior history of that disease; however, relatives and adoptees of someone with that disease are far more likely (but not certain) to develop it, indicating a strong genetic component. The liability threshold model was developed to deal with these non-Mendelian binary cases; the model proposes that there is a continuous normally-distributed trait expressing risk polygenically influenced by many genes, which all individuals above a certain value develop the disease and all below it do not.
The first threshold models in genetics were introduced by Sewall Wright, examining the propensity of guinea pig strains to have an extra hind toe, a phenomenon which could not be explained as a dominant or recessive gene, or continuous "blinding inheritance".[7][8] The modern liability-threshold model was introduced into human research by geneticist Douglas Scott Falconer in his textbook[9] and two papers.[10][11] Falconer had been asked about the topic of modeling 'threshold characters' by Cyril Clarke who had diabetes.[12]
An early application of liability-threshold models was to schizophrenia by Irving Gottesman & James Shields, finding substantial heritability & little shared-environment influence[13] and undermining the "cold mother" theory of schizophrenia.
Global boiling
The proposition that global temperature will rise in a non-linear mode once it crosses a hypothetical threshold value has been made in several studies [14] A recent threshold model [15] predicts that in this suprathreshold state temperature rise will be dramatically sharp and non-graded.
↑ Swaminathan, R. et al. (2021) The Physical Climate at Global Warming Thresholds as Seen in the U.K. Earth System Model , {{DOI: https://doi.org/10.1175/JCLI-D-21-0234.1 }}
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.