
In computer science, a binary search tree (BST), also called an ordered or sorted binary tree, is a rooted binary tree data structure with the key of each internal node being greater than all the keys in the respective node's left subtree and less than the ones in its right subtree. The time complexity of operations on the binary search tree is directly proportional to the height of the tree.

In computer science, a binary tree is a k-ary tree data structure in which each node has at most two children, which are referred to as the left child and the right child. A recursive definition using just set theory notions is that a (non-empty) binary tree is a tuple (L, S, R), where L and R are binary trees or the empty set and S is a singleton set containing the root. Some authors allow the binary tree to be the empty set as well.
In biology, phylogenetics is the study of the evolutionary history and relationships among or within groups of organisms. These relationships are determined by phylogenetic inference methods that focus on observed heritable traits, such as DNA sequences, protein amino acid sequences, or morphology. The result of such an analysis is a phylogenetic tree—a diagram containing a hypothesis of relationships that reflects the evolutionary history of a group of organisms.
A splay tree is a binary search tree with the additional property that recently accessed elements are quick to access again. Like self-balancing binary search trees, a splay tree performs basic operations such as insertion, look-up and removal in O(log n) amortized time. For random access patterns drawn from a non-uniform random distribution, their amortized time can be faster than logarithmic, proportional to the entropy of the access pattern. For many patterns of non-random operations, also, splay trees can take better than logarithmic time, without requiring advance knowledge of the pattern. According to the unproven dynamic optimality conjecture, their performance on all access patterns is within a constant factor of the best possible performance that could be achieved by any other self-adjusting binary search tree, even one selected to fit that pattern. The splay tree was invented by Daniel Sleator and Robert Tarjan in 1985.

In computer science, a tree is a widely used abstract data type that represents a hierarchical tree structure with a set of connected nodes. Each node in the tree can be connected to many children, but must be connected to exactly one parent, except for the root node, which has no parent. These constraints mean there are no cycles or "loops", and also that each child can be treated like the root node of its own subtree, making recursion a useful technique for tree traversal. In contrast to linear data structures, many trees cannot be represented by relationships between neighboring nodes in a single straight line.

A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time. In another word, it is a branching diagram or a tree showing the evolutionary relationships among various biological species or other entities based upon similarities and differences in their physical or genetic characteristics. All life on Earth is part of a single phylogenetic tree, indicating common ancestry. Phylogenetics is the field of the study for the phylogenetic trees. The main challenge is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of species or taxa. Computational phylogenetics focuses on the algorithms involved in finding optimal phylogenetic tree in the phylogenetic landscape.
In phylogenetics and computational phylogenetics, maximum parsimony is an optimality criterion under which the phylogenetic tree that minimizes the total number of character-state changes. Under the maximum-parsimony criterion, the optimal tree will minimize the amount of homoplasy. In other words, under this criterion, the shortest possible tree that explains the data is considered best. Some of the basic ideas behind maximum parsimony were presented by James S. Farris in 1970 and Walter M. Fitch in 1971.
Computational phylogenetics focuses on computational algorithms and approaches involved in phylogenetic analyses. The goal is to find a phylogenetic tree representing optimal evolutionary ancestry between a set of genes, species, or taxa. Maximum likelihood, parsimony, Bayesian, and minimum evolution are typical optimality criteria used to assess how well a phylogenetic tree topology describes the sequence data. Nearest Neighbour Interchange (NNI), Subtree Prune and Regraft (SPR), and Tree Bisection and Reconnection (TBR), known as tree rearrangements, are deterministic algorithms to search for optimal or the best phylogenetic tree. The space and the landscape of searching for the optimal phylogenetic tree is known as phylogeny search space.
Ancestral reconstruction is the extrapolation back in time from measured characteristics of individuals to their common ancestors. It is an important application of phylogenetics, the reconstruction and study of the evolutionary relationships among individuals, populations or species to their ancestors. In the context of evolutionary biology, ancestral reconstruction can be used to recover different kinds of ancestral character states of organisms that lived millions of years ago. These states include the genetic sequence, the amino acid sequence of a protein, the composition of a genome, a measurable characteristic of an organism (phenotype), and the geographic range of an ancestral population or species. This is desirable because it allows us to examine parts of phylogenetic trees corresponding to the distant past, clarifying the evolutionary history of the species in the tree. Since modern genetic sequences are essentially a variation of ancient ones, access to ancient sequences may identify other variations and organisms which could have arisen from those sequences. In addition to genetic sequences, one might attempt to track the changing of one character trait to another, such as fins turning to legs.
In mathematics, Newick tree format is a way of representing graph-theoretical trees with edge lengths using parentheses and commas. It was adopted by James Archie, William H. E. Day, Joseph Felsenstein, Wayne Maddison, Christopher Meacham, F. James Rohlf, and David Swofford, at two meetings in 1986, the second of which was at Newick's restaurant in Dover, New Hampshire, US. The adopted format is a generalization of the format developed by Meacham in 1984 for the first tree-drawing programs in Felsenstein's PHYLIP package.
A tango tree is a type of binary search tree proposed by Erik D. Demaine, Dion Harmon, John Iacono, and Mihai Pătrașcu in 2004. It is named after Buenos Aires, of which the tango is emblematic.
Distance matrices are used in phylogeny as non-parametric distance methods and were originally applied to phenetic data using a matrix of pairwise distances. These distances are then reconciled to produce a tree. The distance matrix can come from a number of different sources, including measured distance or morphometric analysis, various pairwise distance formulae applied to discrete morphological characters, or genetic distance from sequence, restriction fragment, or allozyme data. For phylogenetic character data, raw distance values can be calculated by simply counting the number of pairwise differences in character states.
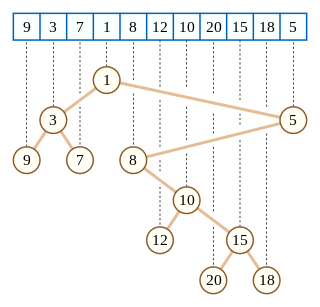
In computer science, a Cartesian tree is a binary tree derived from a sequence of distinct numbers. To construct the Cartesian tree, set its root to be the minimum number in the sequence, and recursively construct its left and right subtrees from the subsequences before and after this number. It is uniquely defined as a min-heap whose symmetric (in-order) traversal returns the original sequence.
Capacitated minimum spanning tree is a minimal cost spanning tree of a graph that has a designated root node and satisfies the capacity constraint . The capacity constraint ensures that all subtrees incident on the root node have no more than nodes. If the tree nodes have weights, then the capacity constraint may be interpreted as follows: the sum of weights in any subtree should be no greater than . The edges connecting the subgraphs to the root node are called gates. Finding the optimal solution is NP-hard.
The Robinson–Foulds or symmetric difference metric, often abbreviated as the RF distance, is a simple way to calculate the distance between phylogenetic trees. It is defined as where A is the number of partitions of data implied by the first tree but not the second tree and B is the number of partitions of data implied by the second tree but not the first tree. The partitions are calculated for each tree by removing each branch. Thus, the number of eligible partitions for each tree is equal to the number of branches in that tree. RF distances have been criticized as biased, but they represent a relatively intuitive measure of the distances between phylogenetic trees and therefore remain widely used. Nevertheless, the biases inherent to the RF distances suggest that researches should consider using "Generalized" Robinson–Foulds metrics that may have better theoretical and practical performance and avoid the biases and misleading attributes of the original metric.

In mathematics and computer science, an unrooted binary tree is an unrooted tree in which each vertex has either one or three neighbors.
In computer science, an optimal binary search tree (Optimal BST), sometimes called a weight-balanced binary tree, is a binary search tree which provides the smallest possible search time (or expected search time) for a given sequence of accesses (or access probabilities). Optimal BSTs are generally divided into two types: static and dynamic.
In computer science, frequent subtree mining is the problem of finding all patterns in a given database whose support is over a given threshold. It is a more general form of the maximum agreement subtree problem.
In the mathematical field of graph theory, an agreement forest for two given trees is any forest which can, informally speaking, be obtained from both trees by removing a common number of edges.

In phylogenetics, reconciliation is an approach to connect the history of two or more coevolving biological entities. The general idea of reconciliation is that a phylogenetic tree representing the evolution of an entity can be drawn within another phylogenetic tree representing an encompassing entity to reveal their interdependence and the evolutionary events that have marked their shared history. The development of reconciliation approaches started in the 1980s, mainly to depict the coevolution of a gene and a genome, and of a host and a symbiont, which can be mutualist, commensalist or parasitic. It has also been used for example to detect horizontal gene transfer, or understand the dynamics of genome evolution.
 Nearest neighbor interchange (NNI)
Nearest neighbor interchange (NNI) Subtree pruning and regrafting (SPR)
Subtree pruning and regrafting (SPR) Tree bisection and reconnection (TBR)
Tree bisection and reconnection (TBR)

