
The United States Food and Drug Administration is a federal agency of the Department of Health and Human Services. The FDA is responsible for protecting and promoting public health through the control and supervision of food safety, tobacco products, caffeine products, dietary supplements, prescription and over-the-counter pharmaceutical drugs (medications), vaccines, biopharmaceuticals, blood transfusions, medical devices, electromagnetic radiation emitting devices (ERED), cosmetics, animal foods & feed and veterinary products.

Over-the-counter (OTC) drugs are medicines sold directly to a consumer without a requirement for a prescription from a healthcare professional, as opposed to prescription drugs, which may be supplied only to consumers possessing a valid prescription. In many countries, OTC drugs are selected by a regulatory agency to ensure that they contain ingredients that are safe and effective when used without a physician's care. OTC drugs are usually regulated according to their active pharmaceutical ingredient (API) rather than final products. By regulating APIs instead of specific drug formulations, governments allow manufacturers the freedom to formulate ingredients, or combinations of ingredients, into proprietary mixtures.
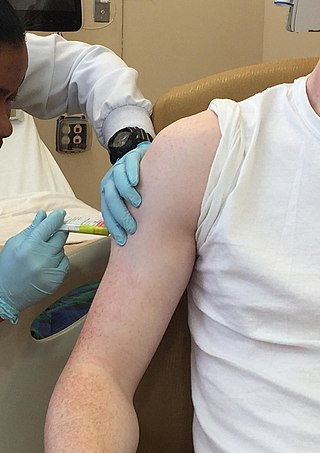
Clinical trials are prospective biomedical or behavioral research studies on human participants designed to answer specific questions about biomedical or behavioral interventions, including new treatments and known interventions that warrant further study and comparison. Clinical trials generate data on dosage, safety and efficacy. They are conducted only after they have received health authority/ethics committee approval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial—their approval does not mean the therapy is 'safe' or effective, only that the trial may be conducted.

A generic drug is a pharmaceutical drug that contains the same chemical substance as a drug that was originally protected by chemical patents. Generic drugs are allowed for sale after the patents on the original drugs expire. Because the active chemical substance is the same, the medical profile of generics is equivalent in performance compared to their performance at the time when they were patented drugs. A generic drug has the same active pharmaceutical ingredient (API) as the original, but it may differ in some characteristics such as the manufacturing process, formulation, excipients, color, taste, and packaging.

A prescription drug is a pharmaceutical drug that is permitted to be dispensed only to those with a medical prescription. In contrast, over-the-counter drugs can be obtained without a prescription. The reason for this difference in substance control is the potential scope of misuse, from drug abuse to practicing medicine without a license and without sufficient education. Different jurisdictions have different definitions of what constitutes a prescription drug.
The Burzynski Clinic is a clinic selling an unproven cancer treatment, which has been characterized as harmful quackery. It was founded in 1976 and is located in Houston, Texas, in the United States. It offers a form of chemotherapy originally called "antineoplaston therapy" devised by the clinic's founder Stanislaw Burzynski in the 1970s. Antineoplaston is Burzynski's term for a group of urine-derived peptides, peptide derivatives, and mixtures. There is no accepted scientific evidence of benefit from antineoplaston combinations for various diseases, and the Clinic's claimed successes have not been replicated by independent researchers. The therapy has been rebranded in various ways over the years to mirror fashions in medicine, for example as a kind of "immunotherapy". The therapy is administered through the ruse of running a large numbers of clinical trials, which long-time Burzynski lawyer Richard Jaffe has described as "a joke".

The Bayh–Dole Act or Patent and Trademark Law Amendments Act is United States legislation permitting ownership by contractors of inventions arising from federal government-funded research. Sponsored by senators, Birch Bayh of Indiana and Bob Dole of Kansas, the Act was adopted in 1980, is codified at 94 Stat. 3015, and in 35 U.S.C. § 200–212, and is implemented by 37 C.F.R. 401 for federal funding agreements with contractors and 37 C.F.R 404 for licensing of inventions owned by the federal government.

The United States Food and Drug Administration's Investigational New Drug (IND) program is the means by which a pharmaceutical company obtains permission to start human clinical trials and to ship an experimental drug across state lines before a marketing application for the drug has been approved. Regulations are primarily at 21 CFR 312. Similar procedures are followed in the European Union, Japan, and Canada.
Off-label use is the use of pharmaceutical drugs for an unapproved indication or in an unapproved age group, dosage, or route of administration. Both prescription drugs and over-the-counter drugs (OTCs) can be used in off-label ways, although most studies of off-label use focus on prescription drugs.
Direct-to-consumer advertising (DTCA) refers to the marketing and advertising of pharmaceutical products directly to consumers as patients, as opposed to specifically targeting health professionals. The term is synonymous primarily with the advertising of prescription medicines via mass media platforms—most commonly on television and in magazines, but also via online platforms.
Abigail Alliance for Better Access to Developmental Drugs v. von Eschenbach, 495 F.3d 695, cert denied, 552 U.S. 1159 (2008) was resolved in early 2008 when the Supreme Court of the United States declined to hear the appeal. Their refusal left standing the appellate court decision, which said that patients have no right to "a potentially toxic drug with no proven therapeutic benefit."
The Prescription Drug User Fee Act (PDUFA) was a law passed by the United States Congress in 1992 which allowed the Food and Drug Administration (FDA) to collect fees from drug manufacturers to fund the new drug approval process. The Act provided that the FDA was entitled to collect a substantial application fee from drug manufacturers at the time a New Drug Application (NDA) or Biologics License Application (BLA) was submitted, with those funds designated for use only in Center for Drug Evaluation and Research (CDER) or Center for Biologics Evaluation and Research (CBER) drug approval activities. In order to continue collecting such fees, the FDA is required to meet certain performance benchmarks, primarily related to the speed of certain activities within the NDA review process.
Expanded access or compassionate use is the use of an unapproved drug or medical device under special forms of investigational new drug applications (IND) or IDE application for devices, outside of a clinical trial, by people with serious or life-threatening conditions who do not meet the enrollment criteria for the clinical trial in progress.
Priority review is a program of the United States Food and Drug Administration (FDA) to expedite the review process for drugs that are expected to have a particularly great impact on the treatment of a disease. The priority review voucher program is a program that grants a voucher for priority review to a drug developer as an incentive to develop treatments for disease indications with limited profitability.
A drug coupon is a coupon intended to help consumers save money on pharmaceutical drugs. They are offered by drug companies or distributed to consumers via doctors and pharmacists, and most can be obtained online. There are drug coupons for drugs from many categories such as cholesterol, acne, migraine, allergies, etc.
The United States Food and Drug Administration Modernization Act of 1997 (FDAMA) amended the Federal Food, Drug, and Cosmetic Act. This act is related to the regulation of food, drugs, devices, and biological products by the FDA. These changes were made in order to recognize the changes in the way the FDA would be operating in the 21st century. The main focus of this is the acknowledgment in the advancement of technological, trade, and public health complexities.
The Food and Drug Administration is a federal agency of the United States, formed in 1930.
The Drug Quality and Security Act is a law that amended the Federal Food, Drug, and Cosmetic Act to grant the Food and Drug Administration more authority to regulate and monitor the manufacturing of compounded drugs. The bill was written in response to the New England Compounding Center meningitis outbreak that took place in 2012, which killed 64 people. The bill was signed by President Obama on November 27, 2013.
Right-to-try laws are United States state laws and a federal law that were created with the intent of allowing terminally ill patients access to experimental therapies that have completed Phase I testing but have not been approved by the Food and Drug Administration (FDA). Prior to the passage of right to try laws, patients needed FDA approval to use experimental drugs. As of 2018, 41 U.S. states had passed right to try laws. The framers of these laws argue that this allows for individualized treatments that are not permitted under the FDA's current regulatory scheme. The value of these laws was questioned on multiple grounds, including the fact that pharmaceutical manufacturers would have no obligation to provide the therapies being sought. A federal right to try law was passed in May 2018. Very little data is available about the number of patients who have used the right-to-try pathway, but available sources indicate that since the signing of the bill only a handful of patients have used this pathway to access experimental therapies, as most physicians and sponsors prefer the more traditional, FDA approved, Expanded Access route. According to Scott Gottlieb, who served as commissioner of the FDA under President Donald Trump, the FDA had already approved 99% of patient requests for access to experimental drugs, either immediately over the phone or within a few days, prior to the passage of right to try legislation.

Aid Access is a nonprofit organization that provides access to medication abortion by mail to the United States and worldwide. It describes its work as a harm reduction strategy designed to provide safe access to mifepristone and misoprostol for those able to become pregnant in the United States who may not otherwise have access to abortion or miscarriage management services. People are able to manage their own abortion with remote access to a physician and a help-desk for any questions. The website is available in English, Spanish, and Dutch.
