
Tetrahedrane is a hypothetical platonic hydrocarbon with chemical formula C4H4 and a tetrahedral structure. The molecule would be subject to considerable angle strain and has not been synthesized as of 2023. However, a number of derivatives have been prepared. In a more general sense, the term tetrahedranes is used to describe a class of molecules and ions with related structure, e.g. white phosphorus.
In organic chemistry, a sigmatropic reaction is a pericyclic reaction wherein the net result is one sigma bond (σ-bond) is changed to another σ-bond in an intramolecular reaction. In this type of rearrangement reaction, a substituent moves from one part of a π-system to another part with simultaneous rearrangement of the π-system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
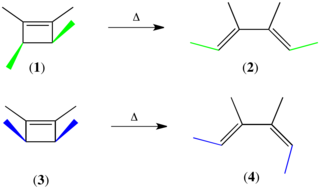
The Woodward–Hoffmann rules are a set of rules devised by Robert Burns Woodward and Roald Hoffmann to rationalize or predict certain aspects of the stereochemistry and activation energy of pericyclic reactions, an important class of reactions in organic chemistry. The rules originate in certain symmetries of the molecule's orbital structure that any molecular Hamiltonian conserves. Consequently, any symmetry-violating reaction must couple extensively to the environment; this imposes an energy barrier on its occurrence, and such reactions are called symmetry-forbidden. Their opposites are symmetry-allowed.

Borirenes are a unique class of three-membered heterocyclic compounds characterized by an unsaturated boron atom within their ring structure. First computationally predicted by John Pople and Paul von Rague Schleyer in 1981, the simplest borirene, (CH)2BH, is isoelectronic with the cyclopropenium cation and exhibits Hückel aromaticity. Borirenes undergo ring-opening reactions with polar reagents and form Lewis adducts, due to the significant ring strain in its three-membered structure and the presence of an empty p orbital on the boron atom. The balance of these two properties leads to unique properties as a ligand for transition metals, in addition to observation of photochemical rearrangement and ring expansion. While borirenes were first discovered in the 1980s, new derivatives such as benzoborirenes have led to renewed interest in the field, with their potential applications yet to be fully explored.

The vinyl cation is a carbocation with the positive charge on an alkene carbon. Its empirical formula of the parent ion is C
2H+
3. Vinyl cation are invoked as reactive intermediates in solvolysis of vinyl halides, as well as electrophilic addition to alkynes and allenes.

Germylenes are a class of germanium(II) compounds with the general formula :GeR2. They are heavier carbene analogs. However, unlike carbenes, whose ground state can be either singlet or triplet depending on the substituents, germylenes have exclusively a singlet ground state. Unprotected carbene analogs, including germylenes, has a dimerization nature. Free germylenes can be isolated under the stabilization of steric hindrance or electron donation. The synthesis of first stable free dialkyl germylene was reported by Jutzi, et al in 1991.

Phosphinidenes are low-valent phosphorus compounds analogous to carbenes and nitrenes, having the general structure RP. The "free" form of these compounds is conventionally described as having a singly-coordinated phosphorus atom containing only 6 electrons in its valence level. Most phosphinidenes are highly reactive and short-lived, thereby complicating empirical studies on their chemical properties. In the last few decades, several strategies have been employed to stabilize phosphinidenes, and researchers have developed a number of reagents and systems that can generate and transfer phosphinidenes as reactive intermediates in the synthesis of various organophosphorus compounds.
Boroles represent a class of molecules known as metalloles, which are heterocyclic 5-membered rings. As such, they can be viewed as structural analogs of cyclopentadiene, pyrrole or furan, with boron replacing a carbon, nitrogen and oxygen atom respectively. They are isoelectronic with the cyclopentadienyl cation C5H+5 or abbreviated as Cp+ and comprise four π electrons. Although Hückel's rule cannot be strictly applied to borole, it is considered to be antiaromatic due to having 4 π electrons. As a result, boroles exhibit unique electronic properties not found in other metalloles.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a (3+2) cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
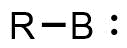
A borylene is the boron analogue of a carbene. The general structure is R-B: with R an organic moiety and B a boron atom with two unshared electrons. Borylenes are of academic interest in organoboron chemistry. A singlet ground state is predominant with boron having two vacant sp2 orbitals and one doubly occupied one. With just one additional substituent the boron is more electron deficient than the carbon atom in a carbene. For this reason stable borylenes are more uncommon than stable carbenes. Some borylenes such as boron monofluoride (BF) and boron monohydride (BH) the parent compound also known simply as borylene, have been detected in microwave spectroscopy and may exist in stars. Other borylenes exist as reactive intermediates and can only be inferred by chemical trapping.
Digermynes are a class of compounds that are regarded as the heavier digermanium analogues of alkynes. The parent member of this entire class is H-Ge≡Ge-H, which has only been characterized computationally, but has revealed key features of the whole class. Because of the large interatomic repulsion between two Ge atoms, only kinetically stabilized digermyne molecules can be synthesized and characterized by utilizing bulky protecting groups and appropriate synthetic methods, for example, reductive coupling of germanium(II) halides.
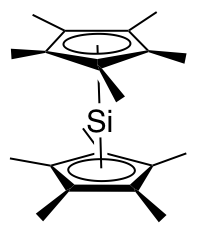
Decamethylsilicocene, (C5Me5)2Si, is a group 14 sandwich compound. It is an example of a main-group cyclopentadienyl complex; these molecules are related to metallocenes but contain p-block elements as the central atom. It is a colorless, air sensitive solid that sublimes under vacuum.

Diphosphagermylenes are a class of compounds containing a divalent germanium atom bound to two phosphorus atoms. While these compounds resemble diamidocarbenes, such as N-heterocyclic carbenes (NHC), diphosphagermylenes display bonding characteristics distinct from those of diamidocarbenes. In contrast to NHC compounds, in which there is effective N-C p(π)-p(π) overlap between the lone pairs of planar nitrogens and an empty p-orbital of a carbene, systems containing P-Ge p(π)-p(π) overlap are rare. Until 2014, the geometry of phosphorus atoms in all previously reported diphosphatetrylenes are pyramidal, with minimal P-Ge p(π)-p(π) interaction. It has been suggested that the lack of p(π)-p(π) in Ge-P bonds is due to the high energetic barrier associated with achieving a planar configuration at phosphorus, which would allow for efficient p(π)-p(π) overlap between the phosphorus lone pair and the empty P orbital of Ge. The resulting lack of π stabilization contributes to the difficulty associated with isolating diphosphagermylene and the Ge-P double bonds. However, utilization of sterically encumbering phosphorus centers has allowed for the isolation of diphosphagermylenes with a planar phosphorus center with a significant P-Ge p(π)-p(π) interaction.

Silylones are a class of zero-valent monatomic silicon complexes, characterized as having two lone pairs and two donor-acceptor ligand interactions stabilizing a silicon(0) center. Synthesis of silylones generally involves the use of sterically bulky carbenes to stabilize highly reactive Si(0) centers. For this reason, silylones are sometimes referred to siladicarbenes. To date, silylones have been synthesized with cyclic alkyl amino carbenes (cAAC) and bidentate N-heterocyclic carbenes (bis-NHC). They are capable of reactions with a variety of substrates, including chalcogens and carbon dioxide.

Phosphasilenes or silylidenephosphanes are a class of compounds with silicon-phosphorus double bonds. Since the electronegativity of phosphorus (2.1) is higher than that of silicon (1.9), the "Si=P" moiety of phosphasilene is polarized. The degree of polarization can be tuned by altering the coordination numbers of the Si and P centers, or by modifying the electronic properties of the substituents. The phosphasilene Si=P double bond is highly reactive, yet with the choice of proper substituents, it can be stabilized via donor-acceptor interaction or by steric congestion.

Tellurophenes are the tellurium analogue of thiophenes and selenophenes.

An N-Heterocyclic silylene (NHSi) is a neutral heterocyclic chemical compound consisting of a divalent silicon atom bonded to two nitrogen atoms. The isolation of the first stable NHSi, also the first stable dicoordinate silicon compound, was reported in 1994 by Michael Denk and Robert West three years after Anthony Arduengo first isolated an N-heterocyclic carbene, the lighter congener of NHSis. Since their first isolation, NHSis have been synthesized and studied with both saturated and unsaturated central rings ranging in size from 4 to 6 atoms. The stability of NHSis, especially 6π aromatic unsaturated five-membered examples, make them useful systems to study the structure and reactivity of silylenes and low-valent main group elements in general. Though not used outside of academic settings, complexes containing NHSis are known to be competent catalysts for industrially important reactions. This article focuses on the properties and reactivity of five-membered NHSis.
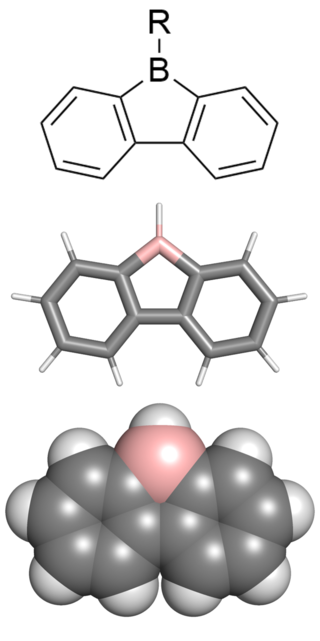
9-borafluorenes are a class of boron-containing heterocycles consisting of a tricyclic system with a central BC4 ring with two fused arene groups. 9-borafluorenes can be thought of as a borole with two fused arene rings, or as a trigonal planar boron atom with an empty p orbital bridging two biphenyl rings. However, 9-borafluorenes are generally less reactive than boroles due to less antiaromatic character and Lewis acidity. Containing highly conjugated π systems, 9-borafluorenes possess interesting photophysical properties. In addition, 9-borafluorenes are good Lewis acids. This combination of properties enables potential uses such as in light-emitting materials, solar cells, and sensors for some molecules.
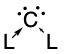
Carbones are a class of molecules containing a carbon atom in the 1D excited state with a formal oxidation state of zero where all four valence electrons exist as unbonded lone pairs. These carbon-based compounds are of the formula CL2 where L is a strongly σ-donating ligand, typically a phosphine (carbodiphosphoranes) or a N-heterocyclic carbene/NHC (carbodicarbenes), that stabilises the central carbon atom through donor-acceptor bonds. Carbones possess high-energy orbitals with both σ- and π-symmetry, making them strong Lewis bases and strong π-backdonor substituents. Carbones possess high proton affinities and are strong nucleophiles which allows them to function as ligands in a variety of main group and transition metal complexes. Carbone-coordinated elements also exhibit a variety of different reactivities and catalyse various organic and main group reactions.
Principal interacting orbital (PIO), based on quantum chemical calculations, provides chemists with visualization of a set of semi-localized dominant interacting orbitals. The method offers additional perspective to molecular orbitals (MO) obtained from quantum chemical calculations, which often provide extensively delocalized orbitals that are hard to interpret and relate with chemists' intuition on electronic structures and orbital interactions. Several other efforts have been made to help visualize semi-localized dominant interacting orbitals that represents well chemists' intuition, while maintaining the mathematical rigorosity. Notable examples include the natural atomic orbitals (NAO), natural bond orbitals (NBO), charge decomposition analysis (CDA), and adaptive natural density partitioning (AdNDP). PIO analysis uniquely provides semi-localized MOs that are chemically accurate and easy to interpret.










