Related Research Articles

Microcephaly is a medical condition involving a smaller-than-normal head. Microcephaly may be present at birth or it may develop in the first few years of life. Brain development is often affected; people with this disorder often have an intellectual disability, poor motor function, poor speech, abnormal facial features, seizures and dwarfism.

Helicases are a class of enzymes thought to be vital to all organisms. Their main function is to unpack an organism's genetic material. Helicases are motor proteins that move directionally along a nucleic acid phosphodiester backbone, separating two hybridized nucleic acid strands, using energy from ATP hydrolysis. There are many helicases, representing the great variety of processes in which strand separation must be catalyzed. Approximately 1% of eukaryotic genes code for helicases.
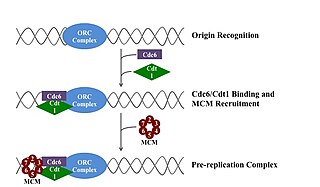
A pre-replication complex (pre-RC) is a protein complex that forms at the origin of replication during the initiation step of DNA replication. Formation of the pre-RC is required for DNA replication to occur. Complete and faithful replication of the genome ensures that each daughter cell will carry the same genetic information as the parent cell. Accordingly, formation of the pre-RC is a very important part of the cell cycle.
RecQ helicase is a family of helicase enzymes initially found in Escherichia coli that has been shown to be important in genome maintenance. They function through catalyzing the reaction ATP + H2O → ADP + P and thus driving the unwinding of paired DNA and translocating in the 3' to 5' direction. These enzymes can also drive the reaction NTP + H2O → NDP + P to drive the unwinding of either DNA or RNA.

Bloom syndrome is a rare autosomal recessive genetic disorder characterized by short stature, predisposition to the development of cancer, and genomic instability. BS is caused by mutations in the BLM gene which is a member of the RecQ DNA helicase family. Mutations in other members of this family, namely WRN and RECQL4, are associated with the clinical entities Werner syndrome and Rothmund–Thomson syndrome, respectively. More broadly, Bloom syndrome is a member of a class of clinical entities that are characterized by chromosomal instability, genomic instability, or both and by cancer predisposition.

Werner syndrome ATP-dependent helicase, also known as DNA helicase, RecQ-like type 3, is an enzyme that in humans is encoded by the WRN gene. WRN is a member of the RecQ Helicase family. Helicase enzymes generally unwind and separate double-stranded DNA. These activities are necessary before DNA can be copied in preparation for cell division. Helicase enzymes are also critical for making a blueprint of a gene for protein production, a process called transcription. Further evidence suggests that Werner protein plays a critical role in repairing DNA. Overall, this protein helps maintain the structure and integrity of a person's DNA.
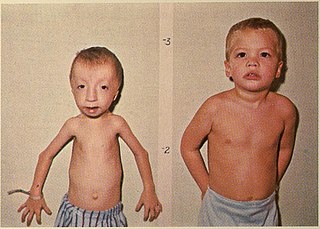
Seckel syndrome, or microcephalic primordial dwarfism is an extremely rare congenital nanosomic disorder. Inheritance is autosomal recessive. It is characterized by intrauterine growth restriction and postnatal dwarfism with a small head, narrow bird-like face with a beak-like nose, large eyes with down-slanting palpebral fissures, receding mandible and intellectual disability.

RAPADILINO syndrome is an autosomal recessive disorder characterized by:

Uncombable hair syndrome (UHS) is a rare structural anomaly of the hair with a variable degree of effect. It is characterized by hair that is silvery, dry, frizzy, wiry, and impossible to comb. It was first reported in the early 20th century. It typically becomes apparent between the ages of 3 months and 12 years. UHS has several names, including "pili trianguli et canaliculi," "cheveux incoiffables," and "spun-glass hair." This disorder is believed to be autosomal recessive in most instances, but there are a few documented cases where multiple family members display the trait in an autosomal dominant fashion. Based on the current scientific studies related to the disorder, the three genes that have been causally linked to UHS are PADI3, TGM3, and TCHH. These genes encode proteins important for hair shaft formation. Clinical symptoms of the disorder arise between 3 months and 12 years of age. The quantity of hair on the head does not change, but hair starts to grow more slowly and becomes increasingly "uncombable." To be clinically apparent, 50% of all scalp hair shafts must be affected by UHS. This syndrome only affects the hair shaft of the scalp and does not influence hair growth in terms of quantity, textural feel, or appearance on the rest of the body.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.

Twinkle protein also known as twinkle mtDNA helicase is a mitochondrial protein that in humans is encoded by the TWNK gene located in the long arm of chromosome 10 (10q24.31).

Probable ATP-dependent RNA helicase DDX11 is an enzyme that in humans is encoded by the DDX11 gene.

LIG4 syndrome is an extremely rare condition caused by mutations in the DNA Ligase IV (LIG4) gene. Some mutations in this gene are associated with a resistance against multiple myeloma and Severe Combined Immunodeficiency. Severity of symptoms depends on the degree of reduced enzymatic activity of Ligase IV or gene expression. Ligase IV is a critical component of the non-homologous end joining (NHEJ) mechanism that repairs DNA double-strand breaks. It is employed in repairing DNA double-strand breaks caused by reactive oxygen species produced by normal metabolism, or by DNA damaging agents such as ionizing radiation. NHEJ is also used to repair the DNA double-strand break intermediates that occur in the production of T and B lymphocyte receptors.

Cernunnos deficiency is a form of combined immunodeficiency characterized by microcephaly, due to mutations in the NHEJ1 gene, it is inherited via autosomal recessive manner Management for this condition is antiviral prophylaxis and antibiotic treatment
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.

RRM3 is a gene that encodes a 5′-to-3′ DNA helicase known affect multiple cellular replication and repair processes and is most commonly studied in Saccharomyces cerevisiae. RRM3 formally stands for Ribosomal DNArecombination mutation 3. The gene codes for nuclear protein Rrm3p, which is 723 amino acids in length, and is part of a Pif1p DNA helicase sub-family that is conserved from yeasts to humans. RRM3 and its encoded protein have been shown to be vital for cellular replication, specifically associating with replication forks genome-wide. RRM3 is located on chromosome 8 in yeast cells and codes for 723 amino acids producing a protein that weighs 81,581 Da.
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.
Infantile cerebral and cerebellar atrophy with postnatal progressive microcephaly is a rare hereditary autosomal recessive malformation syndrome of the central nervous system characterized by profound motor delays and intellectual disabilities, progressive microcephaly, hypertonia, spasticity, clonus and epilepsy. MRI findings include severe cerebellar and cerebral deterioration (atrophy) and impaired myelination. This condition is an example of consequences from the Founder effect, especially that of Jewish populations.
Mandibulofacial dysostosis with microcephaly syndrome, also known as growth delay-intellectual disability-mandibulofacial dysostosis-microcephaly-cleft palate syndrome, mandibulofacial dysostosis, guion-almeida type, or simply as MFDM syndrome is a rare genetic disorder which is characterized by developmental delays, intellectual disabilities, and craniofacial dysmorphisms.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
References
- ↑ "OMIM Entry - # 613398 - WARSAW BREAKAGE SYNDROME; WABS". omim.org. Retrieved 29 October 2019.
- 1 2 Alkhunaizi, Ebba; Shaheen, Ranad; Bharti, Sanjay Kumar; et al. (November 2018). "Warsaw breakage syndrome: Further clinical and genetic delineation". American Journal of Medical Genetics Part A. 176 (11): 2404–2418. doi:10.1002/ajmg.a.40482. PMC 6289708 . PMID 30216658.
- 1 2 Pisani, Francesca M. (5 December 2019). "Spotlight on Warsaw Breakage Syndrome". The Application of Clinical Genetics. 12: 239–248. doi: 10.2147/TACG.S186476 . PMC 6901054 . PMID 31824187.
- 1 2 3 Alkhunaizi, Ebba; Brosh, Robert M.; Alkuraya, Fowzan S.; Chitayat, David (1993). "Warsaw Syndrome". GeneReviews. University of Washington, Seattle. PMID 31169992 . Retrieved 9 April 2023.
- 1 2 3 "Warsaw Breakage Syndrome". GOV.IL. Retrieved 9 April 2023.
- ↑ "DDX11 gene: MedlinePlus Genetics". medlineplus.gov. Retrieved 9 April 2023.
- 1 2 "Warsaw breakage syndrome: MedlinePlus Genetics". medlineplus.gov. Retrieved 9 April 2023.
 This article incorporates text from this source, which is in the public domain .
This article incorporates text from this source, which is in the public domain . - ↑ van der Lelij, Petra; Chrzanowska, Krystyna H.; Godthelp, Barbara C.; Rooimans, Martin A.; Oostra, Anneke B.; Stumm, Markus; Zdzienicka, Małgorzata Z.; Joenje, Hans; de Winter, Johan P. (February 2010). "Warsaw Breakage Syndrome, a Cohesinopathy Associated with Mutations in the XPD Helicase Family Member DDX11/ChlR1". The American Journal of Human Genetics. 86 (2): 262–266. doi:10.1016/j.ajhg.2010.01.008. PMC 2820174 . PMID 20137776.